Protein Misfolding in the Pathogenesis and Diagnosis of Neurodegenerative Diseases (Review)
This review systematizes existing data on protein misfolding in the pathogenesis of neurodegenerative diseases (with a focus on α-synuclein, β-amyloid, and tau protein). Modern laboratory and neuroimaging methods used for clinical diagnosis and scientific research of proteinopathies are discussed. The paper describes promising protein amplification techniques that enable the detection of ultra-low concentrations of aberrant protein forms in biological fluids. The challenges and prospects of early diagnosis of neurodegenerative diseases through protein misfolding detection are also shown.
Introduction
Neurodegenerative diseases (NDDs) are a broad group of pathologies characterized by neuronal death and progressive dysfunction in various nervous system regions, leading to permanent disability in patients [1]. The high prevalence of NDDs, linked to increasing life expectancy, along with significant healthcare expenditures for elderly care, determines the importance of searching for effective methods for early diagnosis of these conditions [2].
It is well-established that the pathogenesis of many NDDs involves protein misfolding and the accumulation of protein fibrils and oligomers [3]. For example, Alzheimer’s disease (AD) is associated with extracellular accumulation of β-amyloid (Aβ) and tau protein in the brain parenchyma. Tau protein aggregation in nervous tissue is also observed in other NDDs, such as frontotemporal dementia (FTD) [4]. Another common NDD, Parkinson’s disease (PD), is characterized by the accumulation of α-synuclein aggregates in neurons, which also underlies dementia with Lewy bodies and (with glial accumulation) multiple system atrophy [5]. Amyotrophic lateral sclerosis, in turn, involves the accumulation of SOD1, FUS, and TDP-43 in motor neurons of the brain [6]. Additionally, prion diseases, classified as spongiform encephalopathies, are accompanied by the deposition of aberrant forms of the PrPC (PrPSC) protein [7, 8].
Currently, the diagnosis of NDDs relies on clinical symptoms, postmortem examination, and neuroimaging techniques [9, 10]. However, it is known that proteinopathy signs can appear years before clinical manifestation and morphological changes in neurodegeneration [11, 12]. Therefore, detecting aberrant proteins in biological fluids represents a promising approach for developing new methods for early NDD diagnosis.
This review studies pathogenetic aspects of protein misfolding (with a focus on α-synuclein and Aβ) as a mechanism of neurodegeneration, as well as modern laboratory and neuroimaging methods used for clinical diagnosis of cerebral proteinopathies. Moreover, the article describes promising protein amplification techniques that enable detection of very low concentrations of aberrant protein forms in biological fluids.
The role of aberrant proteins and their aggregates in the development of neurodegenerative diseases
The most common proteinopathies leading to NDDs involve the accumulation of Aβ, tau, and α-synuclein [13]. Aβ consists of 37–49 amino acid residues and is produced through proteolytic cleavage of the transmembrane amyloid precursor protein (APP) by β- and γ-secretases [13]. APP is expressed in many tissues, including the brain, and is an important regulator of cell proliferation and neurogenesis [14]. One of the pathogenic links in Alzheimer’s-type neurodegeneration is the formation of amyloid plaques (Figure 1), primarily composed of Aβ40/42 peptides [15–17]. There is data showing that Aβ40/42 oligomers are the most toxic compared to other forms [17]. However, the presence of Aβ37, 38, and 40 peptide alloforms in heterogeneous mixtures (e.g., brain interstitial fluid) inhibits the aggregation of the more toxic Aβ42 form [18].
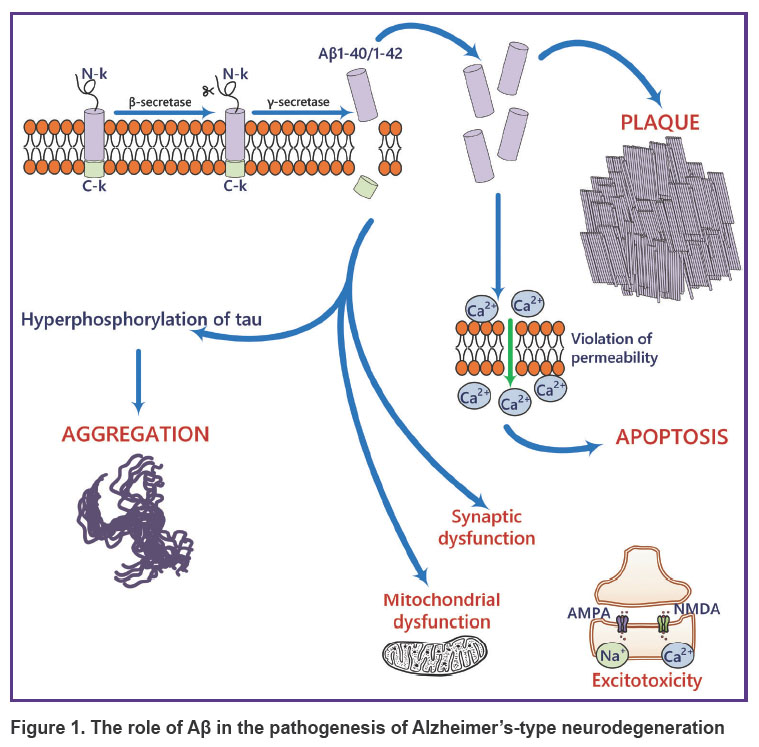
|
Figure 1. The role of Aβ in the pathogenesis of Alzheimer’s-type neurodegeneration |
Tau protein accumulation occurs in various diseases, including AD, FTD, progressive supranuclear palsy, corticobasal degeneration, and Pick’s disease [19, 20]. In healthy neurons, tau is predominantly found in axons; one of its main functions is to stabilize microtubules [21]. Alternative splicing gives rise to different tau isoforms [19]. For example, depending on the presence and number of N-terminal fragments in the molecule, the protein form can be named as 0N, 1N, or 2N. The presence or absence of the R2 domain determines the 4R or 3R forms, respectively [22]. Tauopathies caused by 3R isoform accumulation include Pick’s disease and FTD, while 4R-tau accumulation is observed in corticobasal degeneration and progressive supranuclear palsy. Mixed 3R/4R isoforms are associated with AD and some types of FTD [23, 24]. In NDDs, tau phosphorylation is also shown to stimulate its aggregation and lead to the progression of the disease caused by this protein [25]. Phosphorylated tau also exists in several isoforms (p-tau 181, 217, 231, etc.), which can be used for laboratory diagnosis of NDDs [26, 27].
The presynaptic neuronal protein α-synuclein, which regulates synaptic vesicle movement and further neurotransmitter release, plays a key role in the development of PD and other synucleinopathies (Figure 2) [28]. α-Synuclein consists of 140 amino acids structured into three regions: an N-terminal aliphatic domain (residues 1–60), a hydrophobic domain (residues 61–95), and a C-terminal domain (residues 96–140) [29, 30]. Aggregation of aberrant α-synuclein forms leads to the formation of intracellular inclusions in neurons in PD and dementia with Lewy bodies, as well as glial inclusions in multiple system atrophy [31].
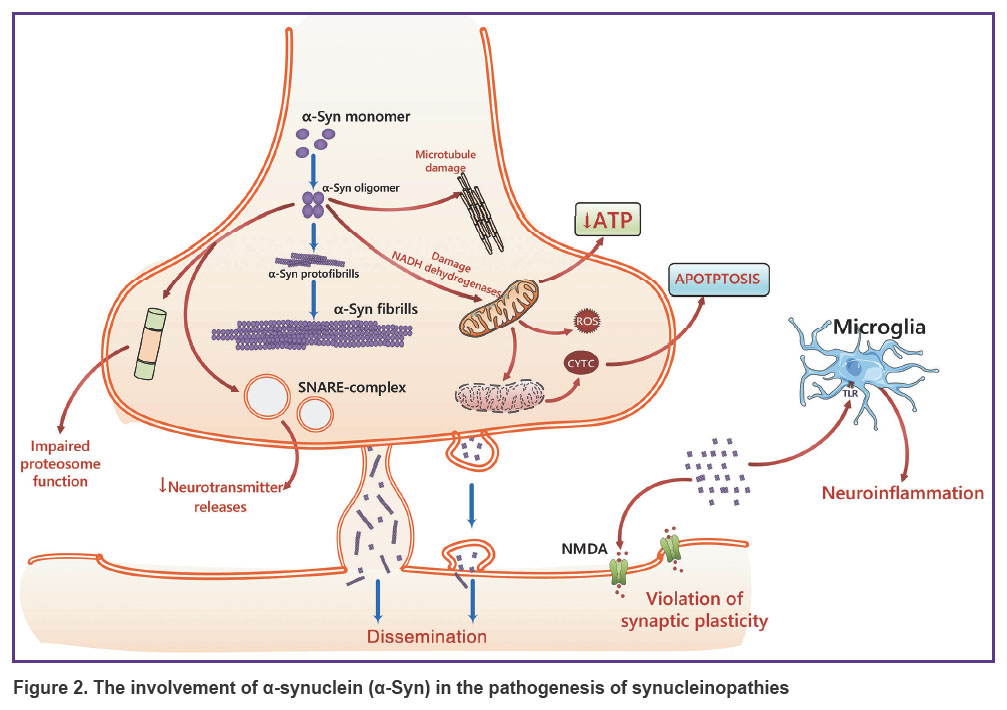
|
Figure 2. The involvement of α-synuclein (α-Syn) in the pathogenesis of synucleinopathies |
Protein misfolding is one of the key pathogenic mechanisms in NDDs, leading to aberrant protein forms and disease progression [32]. Misfolded proteins can adopt various conformations, some of which are prone to aggregation and the formation of highly organized fibrillar structures (amyloidogenesis) [33]. X-ray fiber diffraction has revealed that these proteins are composed of repeating β-sheets arranged antiparallel to each other and perpendicular to the long axis of the fibril [34]. Amyloid fibrils are resistant to detergents and proteases and bind to some dyes such as thioflavin T (ThT) and Congo red, enabling their detection by photometric methods [35].
Protein misfolding and subsequent fibrillogenesis can be facilitated by several factors (e.g., genetic mutations causing alterations in the primary protein structure or in enzymes responsible for protein metabolism) [36]. Specifically, APP gene mutations altering amyloid precursor protein structure, mutations in PSEN1 and PSEN2 genes affecting γ-secretase function, and the APOE-ε4 allele are frequently observed in patients with familial forms of AD [37, 38]. Environmental factors, such as toxins, infections, stress [39, 40], as well as neuroinflammation, oxidative stress, proteostasis system disturbances (autophagy, HSP70/90, ubiquitin), and post-translational protein modifications, predispose to the formation of aberrant protein variants [41–43].
The fibrillogenesis process involves several stages: lag phase, growth phase, and plateau phase [44, 45]. During the lag phase, monomers form a “nucleus” through interactions between special molecule regions called aggregation-prone regions (APRs), primarily composed of hydrophobic amino acids oriented toward the protein core [46, 47]. The elongation phase involves protofibril growth based on previously formed “nuclei” that act as a primer to which monomers attach. The plateau phase is characterized by the depletion of monomers, the cessation of protofibril growth, and the formation of fibrils [45].
Amyloid spread in NDDs often follows specific patterns. For example, in PD, α-synuclein inclusions initially appear in the motor nuclei of the glossopharyngeal and vagus nerves, as well as olfactory tracts, gradually spreading rostrally to higher brain regions [48, 49]. In AD, tau neurofibrillary tangles first appear in the locus coeruleus and transentorhinal cortex, later involving the limbic system and neocortex. Aβ deposits also spread in a distinct pattern, different from the tau pattern, initially accumulating in the neocortex before reaching the hippocampus and deep brain structures [50, 51]. Similar Aβ and tau spread patterns have been observed via positron emission tomography (PET) in AD patients, though there have been observed differences which can be explained by the influence of other factors on the pathological process [52]. This pattern of protein aggregation spread may occur due to the particularly high vulnerability of specific neuronal populations to neurodegenerative processes, which is most likely associated with the difference in some gene expression in neurons, glial cells, or, for example, with the higher energy demands of some neurons [53–55].
According to another point of view, the spread of aberrant proteins may occur through a mechanism similar to that of prion protein spread [56, 57]. This mechanism is characterized by the interaction between the infectious agent, represented by PrPSC being the aberrant form of the prion protein, and the endogenous prion protein PrPS. Such interaction leads to the acquisition of a β-sheet structure by PrPS and its conversion into PrPSC [58]. Indeed, it has been demonstrated that Aβ, α-synuclein, and tau exhibit a tendency to misfold when their structure is rich in β-sheets. It promotes enhanced interaction with normal protein molecules and may serve as a matrix for altering their conformation. These modified proteins can be secreted into the extracellular space and transmitted from cell to cell [59–62].
The addition of recombinant α-synuclein fibrils to primary neuronal cultures results in aggregation of endogenous α-synuclein and neuronal death within 14 days [63]. Different types of α-synuclein vary in their ability to induce protein aggregation [64]. Similar findings have been reported for Aβ [65]. Studies in animal models have shown that in APP/PSEN1 mutant mice, intracerebral injections of brain homogenates from AD patients led to accelerated pathology development not only at the injection site but also in distant brain regions. It demonstrates Aβ’s capacity for transneuronal spread between brain areas [66–68]. The Aβ spread is also influenced by the presence of its post-translationally modified variants in the homogenate, such as AβN3pE and AβpSer8 [69]. Injection of Aβ-containing homogenates into primate brains similarly resulted in progressive deterioration of cognitive and motor function test performance [70].
Intracerebral injections of α-synuclein into the striatum, anterior olfactory nucleus, substantia nigra and other brain regions were similarly characterized by spread of the aberrant protein, development of motor deficits and sleep disturbances [71–73]. Unlike Aβ, which when administered intraperitoneally to mice does not induce neurodegeneration [74], peripheral injection of α-synuclein causes protein aggregation in the brain [75].
Tau protein can induce protein aggregation in anatomically connected regions when injected into the brain [76, 77]. Aβ, tau, and α-synuclein can induce aggregation of other proteins involved in NDDs pathogenesis [78–80]. There is evidence for iatrogenic β-amyloidosis following dura mater transplantation, with Aβ deposits observed specifically in superficial cortical layers [81, 82]. Similar findings have been reported for human growth hormone injections [83, 84]. Thus, Aβ, tau, and α-synuclein can act as “seeds” inducing further aggregation and spread of aberrant proteins.
Another mechanism underlying aberrant protein accumulation involves their spread via extracellular vesicles (EVs) [85]. When EVs isolated from brain tissue of patients with PD with Lewy bodies containing α-synuclein were added to neuronal cultures, they were internalized by neurons [86]. Injection of α-synuclein fibril-containing EVs into rat brains induced further protein aggregation and neuronal death [87]. It is shown that sources of EVs in synucleinopathies include not only neurons but also other cells like microglia [88]. Tau-containing EVs demonstrated the ability to induce protein aggregation both in cell cultures and in vivo [89, 90]. Similarly, in Alzheimer’s neurodegeneration, EVs may serve as a driving mechanism for the Aβ spread [91–93]. Importantly, EVs are promising for being used as NDDs biomarkers [94–96]. A recent study by Kluge et al. [96] has demonstrated a possibility to detect EVs containing aberrant α-synuclein forms in patient serum collected up to 10 years before clinical diagnosis of PD or Lewy body dementia.
Furthermore, aberrant protein “seeds” can spread between cells via tunneling nanotubes (TNTs) [97, 98]. This transport involves both homotypic nanotubes (e.g., between neurons) and heterotypic nanotubes (e.g., between neurons and microglia). On the one hand, this type of aberrant protein transport facilitates pathological protein spreading, while on the other hand, it enables neurons to eliminate protein overload. Additionally, neurons may receive mitochondria from other cells in exchange for proteins, which highlights the potential adaptive role of TNTs [99, 100]. Aberrant protein forms can also be secreted via exocytosis and internalized through receptor-mediated endocytosis [101–104].
Instrumental methods for diagnosing neurodegenerative diseases
Neuroimaging techniques. Neuroimaging methods enable the non-invasive assessment of aberrant protein accumulation, the degree of atrophy in specific brain regions, and the exclusion of other causes of neurological deficits. Structural magnetic resonance imaging accurately evaluates the volume and extent of atrophy in particular brain areas based on different magnetic properties of hydrogen atoms within molecules [105].
Another advanced neuroimaging technique is PET with fluorodeoxyglucose, it allows evaluation of metabolic activity in brain tissues [106]. This method is useful for monitoring patients with mild cognitive impairment who may later develop AD and for differentiating between types of dementia. Despite numerous studies demonstrating the efficacy of PET neuroimaging for diagnosing NDDs, its application is limited due to high costs, technical complexity, and radiation exposure for patients. Besides, both magnetic resonance imaging and PET face challenges regarding diagnostic sensitivity in AD [107, 108].
Biomaterials for early diagnosis of NDDs. Most studies on laboratory diagnosis of NDDs rely on cerebrospinal fluid (CSF) as the primary biological sample [24, 109, 110], its collection requires lumbar puncture. The use of other types of biomaterial will reduce invasiveness and improve diagnostic accessibility. Evidence suggests that aberrant protein forms can be detected in blood years before the clinical onset of PD and AD [96, 111], enabling early screening and prediction of disease manifestation.
Immunological laboratory methods. The enzyme-linked immunosorbent assay (ELISA), based on antigen-antibody interactions for biomolecule detection, is one of the most specific and simple laboratory diagnostic techniques. ELISA is widely used to determine pathological proteins in bodily fluids [109, 112, 113]. For example, the Aβ42/Aβ40 ratio is employed to confirm amyloid pathology in AD [114]. This method can also detect phosphorylated forms of tau in CSF [115].
However, although ELISA is routinely used to measure tau levels in CSF and plasma, it is rarely applied to assess tau accumulation in the brains of AD patients [116]. A number of studies have used ELISA to evaluate tau accumulation in relation to disease stage, brain regions, and AD-related changes [117]. Findings indicate that this diagnostic method can reflect the pattern of tau spread across brain areas. Notably, ELISA employs antibodies targeting the late-middle and C-terminal regions of tau, providing a more accurate representation of its neuropathological accumulation and enabling biochemical quantification of tau levels in the brain [118].
The microfluidic fluorescence assay, the enzyme-linked luminescent assay (ELLA), is based on a microfluidic cartridge platform widely used for the quantitative determination of soluble biomarkers [119]. This approach has been applied in some studies to detect NDDs in patients by measuring levels of neurofilament light chains [120].
Single molecule array (SiMoA) is another fluorescence-based method for neurofilament detection, utilizing two highly specific, non-competing monoclonal antibodies and microelements that can isolate and detect individual molecules connected to paramagnetic beads [119]. Given its technological design, SiMoA is stated to have significantly higher sensitivity than traditional ELISA in NDDs diagnostics. Research by Truffi et al. [120] has demonstrated the comparative efficacy of SiMoA and ELLA platforms in detecting neurofilament light chains, highlighting its potential for NDDs diagnosis.
Mass spectrometric analysis. Mass spectrometry is widely used in analytical chemistry and laboratory diagnostics for qualitative and quantitative determination of chemical substances in samples, including the detection of aberrant proteins for NDDs diagnosis [119]. Currently, mass spectrometric methods are more commonly used in discovery and validation of NDDs biomarkers for research rather than clinical practice. Two complementary mass spectrometry approaches exist: large-scale proteomics for biomarker screening and high-cost targeted methods for identifying specific biomarkers [119].
Another proteomic approach for biomarker detection is a surface-enhanced laser desorption/ionization (SELDI/MALDI) mass spectrometry, developed to analyze high-molecular-weight biomolecules (peptides and proteins) [121]. Proteomic analysis makes it possible to assess levels of up to 10,000 individual proteins in a single sample and track their concentration changes for diagnostic and disease-monitoring purposes [122]. Large-scale mass spectrometry studies have characterized proteomes from biological fluids and brain tissues of patients with various NDDs, including AD, PD, FTD, dementia with Lewy bodies, and amyotrophic lateral sclerosis [123–126]. This method allows precise characterization of protein profile, comprising both normal and aberrant proteins, during NDDs progression, as well as selection of effective screening and diagnostic laboratory algorithms.
Diagnostic methods based on the protein misfolding detection
Raman spectroscopy. In the past decade, Raman microspectroscopy was shown as an effective tool for diagnosing NDDs by detecting structural differences between functional and pathological amyloid structures [127, 128]. However, attempts to find a unique Raman spectral signature for beta-amyloid plaques in brain tissue from AD patients were unsuccessful, with no specific spectrum for Aβ plaques identified even after rigorous removal of potential spectral interferences [129].
Cennamo et al. [130] have successfully used surface-enhanced Raman scattering (SERS), being a modified Raman spectroscopy technique using gold or silver nanoparticles to amplify signals, to detect spectral differences in tear fluid from AD patients. Similarly, Carlomagno et al. [131] have demonstrated promising results using saliva for signal enhancement. There is data showing the efficacy of this method for PD diagnosis when combined with microfluidic platforms [132].
Infrared (IR) spectroscopy. IR spectroscopy registers the absorption of infrared radiation by samples. This method has been successfully applied to study EVs isolated from blood and containing aberrant proteins, revealing differences between AD patients and controls [133, 134]. IR spectroscopy has also effectively differentiated AD from other NDDs (PD, FTD, etc.) [135].
Microspectroscopy (μFTIR), a variant of IR spectroscopy, has been used alongside other methods (Raman spectroscopy and immunofluorescence) to identify astrogliosis associated with Aβ plaques in AD brains [136–138].
Electron microscopy. Conventional transmission electron microscopy visualizes amyloid fibrils with high resolution in dried or hydrated states, providing insights into fibril morphology and protofibrillar structures [139]. However, its limitations include complex sample preparation that may alter native protein matrix and fibril structure of the amyloid [112].
Cryo-electron microscopy (cryo-EM), performed at ultra-low temperatures, has revealed that Aβ fibrils from AD patient brain tissue are not only polymorphic, but also share common structural features in peptide organization and protofilament assembly [140]. Notably, these fibril structures, observed by the authors, differed significantly from those already known and formed in vitro/ex vivo [140, 141]. Guerrero-Ferreira et al. [113] used cryo-EM to identify two novel polymorphic structures of full-length human α-synuclein fibrils in Lewy body samples. These data can be considered a clear indication that the fibrillar structures of aberrant proteins in the brain parenchyma in vivo have structural and probably physico-chemical differences that are important to take into account when interpreting results from in vitro/in silico models.
It is worth mentioning that complementary techniques like Fourier-transform infrared spectroscopy, circular dichroism, and oriented circular dichroism are widely used for analyzing pathological cerebral amyloids [142, 143]. These methods help determine the exact secondary structure composition of β-sheet-rich amyloid aggregates and assess subtle structural changes in aberrant proteins and their interactions with lipid bilayer of the membrane [144]. Methods based on circular dichroism can distinguish parallel and antiparallel β-sheet arrangements, therefore, they provide complementary data to spectroscopy and electron microscopy [145].
Protein amplification techniques. Special attention should be given to protein amplification methods, which detect aberrant proteins in samples of tissues and fluids at ultra-low concentrations. It offers broad diagnostic potential [146]. As mentioned earlier, aberrant proteins can induce misfolding of their native monomers, which underlies the development of prion diseases and NDDs [8, 82]. Amplification techniques are based on this principle, achieving exceptional sensitivity (up to 10–12 g/ml) and specificity (up to 100%) [147, 148] for aberrant protein detection. Given that amyloidogenic protein fragments can appear in biological fluids (blood, CSF, saliva) long before clinical symptoms [11, 95, 96], protein amplification methods are promising as priority approaches for NDDs screening and early diagnosis [96].
One of the earliest and most widely used protein amplification protocols is PMCA (protein misfolding cyclic amplification). In this method, an analyte containing prion-like protein oligomers (PrРSC) is incubated with material containing an excess of normal protein monomers (PrРC), inducing their conformational changes and polymerization (elongation phase) [146]. The resulting polymeric fibrils are cyclically disrupted by ultrasound, increasing the number of PrPSC monomers available for interaction with PrРC, leading to accumulation of PrPSC fibrils. Subsequent detection of the aberrant protein is performed by Western blotting [149]. Despite being methodologically complex, time-consuming and labor-intensive, PMCA significantly surpasses immunodiagnostic methods in sensitivity and enables detection of prion-like proteins even when only a single oligomer is present in the sample [150, 151].
Later, the Western blotting method was replaced with thioflavin-based fluorescent detection of fibrils, eliminating the usage of proteinase C, while ultrasonication was replaced with shaking. The modified method was named RT-QuIC (real-time quaking-induced conversion) [147, 152, 153]. Both PMCA and RT-QuIC have shown high efficacy in diagnosing various prion diseases [8, 151, 154, 155], and these methods currently hold great potential for diagnosing other proteinopathies and NDDs [147, 156]. Modified versions of RT-QuIC using silicon beads (0.8–1.0 mm diameter) can accelerate α-synuclein detection in CSF samples to 1–2 days while enabling quantitative measurement of its concentration [157].
Several authors use the term SAA (seed amplification assay) to describe the combination of PMCA and RT-QuIC techniques. SAA has proven to be an effective tool for diagnosing NDDs associated with accumulation of synuclein [147, 158–160], as well as of Aβ and tau [161]. For example, CSF analysis for α-synuclein using this method can identify patients with synucleinopathies with high sensitivity and specificity (88 and 95%, respectively) [159]. High specificity of SAA has also been demonstrated for other NDDs (AD, corticobasal degeneration, progressive supranuclear palsy), though the simultaneous presence of different aberrant proteins has been shown to reduce accuracy [162–164]. It should be noted that for diagnosing synucleinopathies, PMCA and RT-QuIC typically do not employ ultrasonication; PMCA uses shaking instead, while RT-QuIC uses shaking with beads [165].
For multiple system atrophy, SAA demonstrated sensitivity and specificity of 57 and 96%, respectively, which was lower than for other synucleinopathies. This may be due to structural differences in α-synuclein and the use of different buffers and protocols [166]. Lower diagnostic efficacy of SAA was also noted when identifying patients with certain genetically determined forms of PD (PRKN, LRRK2 genes) [167, 168].
While most protein amplification protocols use CSF as the test material, other biological samples can also be employed. For instance, RT-QuIC analysis of nasal mucosa samples from PD patients has shown diagnostic value [169]. Combined analysis of CSF and nasal mucosa scrapings may be more effective for detecting dementia with Lewy bodies, including at preclinical stages [170]. Another study across two different laboratories used nasal mucosa analysis to diagnose PD and multiple system atrophy [171]. The cerebellar subtype of multiple system atrophy, unlike the parkinsonian subtype, did not show protein aggregation in RT-QuIC. However, nasal mucosa demonstrates lower sensitivity and specificity (45.2 and 89.8%, respectively) compared to CSF [171] or samples from olfactory neuron-rich areas [169, 172].
Kuang et al. [173] reported diagnostic efficacy of SAA methods when analyzing skin samples from PD patients (90% sensitivity and 92% specificity). Several authors suggest that blood and saliva could serve as materials for early PD diagnosis. Wang’s research group [174] has demonstrated that combined analysis of these fluids shows particular promise for PD diagnosis. Kluge et al. [96] have found that blood analysis for α-synuclein using extracellular vesicles isolated with NCAM-1 antibodies can identify patients with PD and dementia with Lewy bodies. Moreover, as mentioned earlier, α-synuclein aggregation can be detected up to 10 years before clinical diagnosis.
Another group of NDDs where protein misfolding detection methods show diagnostic potential are tauopathies: Pick’s disease, chronic traumatic encephalopathy, AD, and others [175]. Detection of aberrant tau “seeds” in CSF and blood samples, similar to α-synuclein detection, could simplify diagnosis of these conditions [175]. However, the study of tau is complicated by subtle conformational differences between various NDDs resulting from alternative splicing. Therefore, some studies emphasize the need to use amplification substrates containing identical protein variants involved in the pathogenesis of specific diseases [176–178].
Protein amplification protocols have been developed for detecting tau in 3R-tauopathies (Pick’s disease) using K19CF monomers, particularly the 3R isoform of recombinant tau [176]. These monomers enabled detection of aberrant tau “seeds” in brain samples from Pick’s disease patients at dilutions up to 10–7–10–9 [176]. Similarly, in 4R-tauopathies (corticobasal degeneration, progressive supranuclear palsy), detection of aberrant protein in postmortem CSF samples was achieved with as little as 0.3 fg of protein per 12 ml sample [177]. Analysis of antemortem CSF showed positive results in 69% of progressive supranuclear palsy cases and 50% of corticobasal degeneration cases [177].
The RT-QuIC method demonstrated greater specificity and sensitivity at higher dilutions (10–7–10–10 for AD versus 10–2–10–6 for other NDDs) when diagnosing 4R/3R-tauopathies in postmortem brain samples [178]. Tennant et al. [179] observed that using 4R/3R recombinant tau monomers as amplification substrates resulted in aggregation of aberrant tau across all tauopathy variants. Thus, 4R/3R “seed” monomers may be suitable for diagnosing all types of tauopathies.
In a study by Salvadores et al. [180], PMCA detected Aβ in CSF samples from AD patients at concentrations as low as 3 fmol/ml (90% sensitivity, 92% specificity). Furthermore, PMCA successfully differentiated AD from other neurological disorders and NDDs [180]. Notably, PMCA can quantify Aβ levels following pharmacological interventions, demonstrating its potential for therapy monitoring. For example, Estrada et al. [181] used protein amplification to show reduced Aβ oligomer levels in plasma from rats treated with imatinib (a c-Abl kinase inhibitor) compared to controls.
While PMCA and RT-QuIC remain effective protein amplification research methods, there exist more modern SAA modifications that, in terms of methodological characteristics, may surpass established methods for detecting protein misfolding. For instance, a protein amplification principle underlies the basis of MDS (multimer detection system) method, being a modified ELISA format. MDS method specifically detects multimeric protein forms, significantly enhancing sensitivity [182, 183]. Another modification is RT-FAST that utilizes nanotubes for protein aggregate detection and amplification within 90 min after test initiation [184, 185]. This represents a major improvement over PMCA and RT-QuIC which require at least several dozen hours for detection. Besides, RT-FAST also enables protein quantification and reduces recombinant protein requirements, lowering costs [184, 185].
Nano-QuIC is a modification of the RT-QuIC method. It incorporates metal nanoparticles that interact with surrounding biomolecules through both protein corona formation and direct effects on protein aggregation [186–188]. Christenson et al. [186] demonstrated that adding 50 nm silicon nanoparticles to RT-QuIC protocols reduced detection time by 2.5-fold and increased specificity 10-fold for Creutzfeldt–Jakob disease diagnosis.
A comprehensive comparison of protein amplification methods, including information about their sensitivity and specificity for various NDDs, is provided in referenced Table [164, 165, 166, 180, 189].
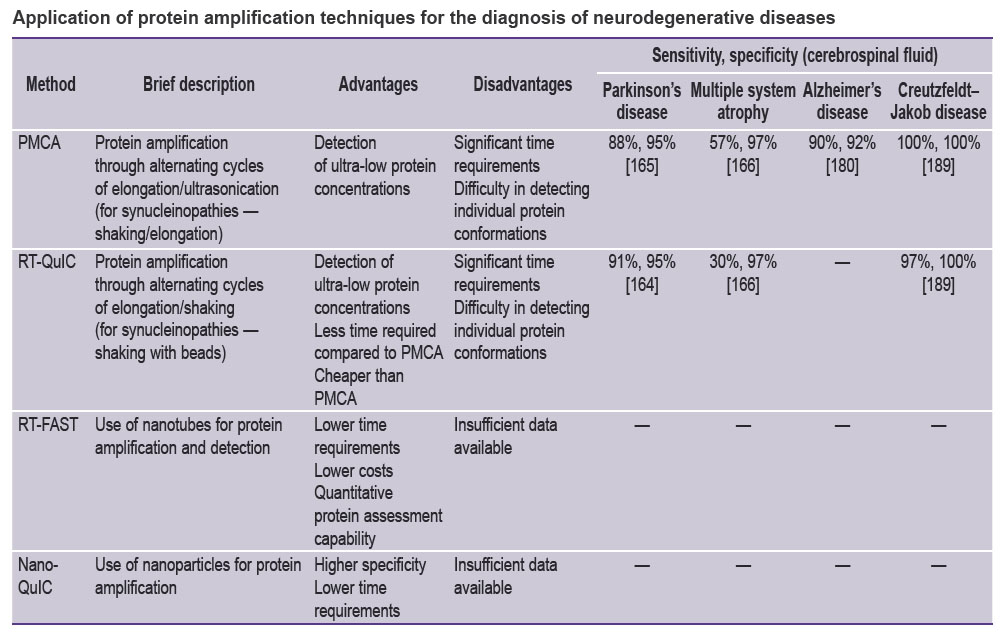
|
Application of protein amplification techniques for the diagnosis of neurodegenerative diseases |
Conclusion
Despite the variety of laboratory and instrumental methods that can be used in the diagnosis of NDDs, not all of them are used in clinical practice due to a number of problems and limitations. Firstly, the pathogenesis of proteinopathies in NDDs has not been sufficiently studied, making it difficult to identify specific biomarkers and their physico-chemical conformations for specific disease. Disorders characterized by the development of parkinsonism have similar symptoms and are characterized by the deposition of different α-synuclein conformations [190]. Secondly, known biomarkers are not always sufficiently specific and may be observed not only in NDDs but also in normal conditions. For example, deposition of Aβ and tau is detected during normal brain aging without neurodegeneration signs [191]. Yet it cannot be ruled out that in such cases there is an early stage of proteinopathy occurring long before the clinical manifestation of the disease.
Another limitation for the application of laboratory methods for diagnosing proteinopathies is the necessity to use CSF as a biological material, since it contains the highest concentrations of aberrant proteins detectable by routine immunological methods [24]. CSF collection is accompanied by a complex, invasive procedure with a number of contraindications, which makes it impossible to expand diagnostic indications and screening.
Despite their technological and methodological complexity, the introduction and development of methods based on the aberrant protein amplification is a promising field in the laboratory diagnosis of NDDs. These methods may enable the detection of ultra-low concentrations of pathological protein conformations in biological bodily fluids long before the clinical onset. However, currently these methods have such disadvantages as long analysis time (up to 7–14 days), low portability, and difficulties in detecting individual conformations of aberrant protein. In addition, a significant limitation for the widespread implementation of protein misfolding detection methods is the complex and labor-intensive process of synthesizing specific monomers used as substrates for amplification.
In this regard, the search for new approaches to develop methods for detecting protein misfolding is relevant. These include, for example, combining microfluidic technologies with the SAA method (PMCA + RT-QuIC), which helps ensure portability, reduce the consumption of components and materials, and significantly accelerate the analysis time [192–194]. Another approach to improve the diagnostic efficiency and technical-economic characteristics of SAA methods may be the application of additional external effects in the protein amplification procedure, such as physical influences (electric and magnetic fields) and the addition of nanoparticles with various properties. These modifications could hypothetically affect the process of oligomer destruction, increasing the number of aberrant protein “seeds” available for amplification.
Study funding. The work was supported by a grant from the Ministry of Science and Higher Education of the Russian Federation for the implementation of large-scale scientific projects in priority areas of scientific and technological development (Project No.075-15-2024-638).
Conflict of interest. The authors declare no conflicts of interest.
References
- Agnello L., Ciaccio M. Neurodegenerative diseases: from molecular basis to therapy. Int J Mol Sci 2022; 23(21): 12854, https://doi.org/10.3390/ijms232112854.
- Feigin V.L., Vos T., Nichols E., Owolabi M.O., Carroll W.M., Dichgans M., Deuschl G., Parmar P., Brainin M., Murray C. The global burden of neurological disorders: translating evidence into policy. Lancet Neurol 2020; 19(3): 255–265, https://doi.org/10.1016/S1474-4422(19)30411-9.
- Jan A., Gokce O., Luthi-Carter R., Lashuel H.A. The ratio of monomeric to aggregated forms of Abeta40 and Abeta42 is an important determinant of amyloid-beta aggregation, fibrillogenesis, and toxicity. J Biol Chem 2008; 283(42): 28176–28189, https://doi.org/10.1074/jbc.M803159200.
- Zhang Y., Wu K.M., Yang L., Dong Q., Yu J.T. Tauopathies: new perspectives and challenges. Mol Neurodegener 2022; 17(1): 28, https://doi.org/10.1186/s13024-022-00533-z.
- Bordbar S., Alijanzadeh D., Samieefar N., Khazeei Tabari M.A., Pourbakhtyaran E., Rezaei N. The role of alpha-synuclein in neurodevelopmental diseases. Mol Neurobiol 2025; 62(1): 962–972, https://doi.org/10.1007/s12035-024-04305-2.
- Sharma R., Khan Z., Mehan S., Das Gupta G., Narula A.S. Unraveling the multifaceted insights into amyotrophic lateral sclerosis: genetic underpinnings, pathogenesis, and therapeutic horizons. Mutat Res Rev Mutat Res 2024; 794: 108518, https://doi.org/10.1016/j.mrrev.2024.108518.
- Calabrese G., Molzahn C., Mayor T. Protein interaction networks in neurodegenerative diseases: from physiological function to aggregation. J Biol Chem 2022; 298(7): 102062, https://doi.org/10.1016/j.jbc.2022.102062.
- Li B., Chen M., Zhu C. Neuroinflammation in prion disease. Int J Mol Sci 2021; 22(4): 2196, https://doi.org/10.3390/ijms22042196.
- Pérez-Millan A., Thirion B., Falgàs N., Borrego-Écija S., Bosch B., Juncà-Parella J., Tort-Merino A., Sarto J., Augé J.M., Antonell A., Bargalló N., Balasa M., Lladó A., Sánchez-Valle R., Sala-Llonch R. Beyond group classification: probabilistic differential diagnosis of frontotemporal dementia and Alzheimer’s disease with MRI and CSF biomarkers. Neurobiol Aging 2024; 144: 1–11, https://doi.org/10.1016/j.neurobiolaging.2024.08.008.
- Erkkinen M.G., Kim M.O., Geschwind M.D. Clinical neurology and epidemiology of the major neurodegenerative diseases. Cold Spring Harb Perspect Biol 2018; 10(4): a033118, https://doi.org/10.1101/cshperspect.a033118.
- Magalhães P., Lashuel H.A. Opportunities and challenges of alpha-synuclein as a potential biomarker for Parkinson’s disease and other synucleinopathies. NPJ Parkinsons Dis 2022; 8(1): 93, https://doi.org/10.1038/s41531-022-00357-0.
- Jia J., Ning Y., Chen M., Wang S., Yang H., Li F., Ding J., Li Y., Zhao B., Lyu J., Yang S., Yan X., Wang Y., Qin W., Wang Q., Li Y., Zhang J., Liang F., Liao Z., Wang S. Biomarker changes during 20 years preceding Alzheimer’s disease. N Engl J Med 2024; 390(8): 712–722, https://doi.org/10.1056/NEJMoa2310168.
- Tatarnikova O.G., Orlov M.A., Bobkova N.V. Beta-amyloid and tau-protein: structure, interaction, and prion-like properties. Biochemistry (Mosc) 2015; 80(13): 1800–1819, https://doi.org/10.1134/S000629791513012X.
- Arber C., Lovejoy C., Harris L., Willumsen N., Alatza A., Casey J.M., Lines G., Kerins C., Mueller A.K., Zetterberg H., Hardy J., Ryan N.S., Fox N.C., Lashley T., Wray S. Familial Alzheimer’s disease mutations in PSEN1 lead to premature human stem cell neurogenesis. Cell Rep 2021; 34(2): 108615, https://doi.org/10.1016/j.celrep.2020.108615.
- Li Y., Schindler S.E., Bollinger J.G., Ovod V., Mawuenyega K.G., Weiner M.W., Shaw L.M., Masters C.L., Fowler C.J., Trojanowski J.Q., Korecka M., Martins R.N., Janelidze S., Hansson O., Bateman R.J. Validation of plasma amyloid-β 42/40 for detecting Alzheimer disease amyloid plaques. Neurology 2022; 98(7): e688–e699, https://doi.org/10.1212/WNL.0000000000013211.
- Yu H., Wu J. Amyloid-β: a double agent in Alzheimer’s disease? Biomed Pharmacother 2021; 139: 111575, https://doi.org/10.1016/j.biopha.2021.111575.
- Sulatskaya A.I., Rychkov G.N., Sulatsky M.I., Mikhailova E.V., Melnikova N.M., Andozhskaya V.S., Kuznetsova I.M., Turoverov K.K. New evidence on a distinction between Aβ40 and Aβ42 amyloids: thioflavin t binding modes, clustering tendency, degradation resistance, and cross-seeding. Int J Mol Sci 2022; 23(10): 5513, https://doi.org/10.3390/ijms23105513.
- Braun G.A., Dear A.J., Sanagavarapu K., Zetterberg H., Linse S. Amyloid-β peptide 37, 38 and 40 individually and cooperatively inhibit amyloid-β 42 aggregation. Chem Sci 2022; 13(8): 2423–2439, https://doi.org/10.1039/d1sc02990h.
- Goedert M., Eisenberg D.S., Crowther R.A. Propagation of tau aggregates and neurodegeneration. Annu Rev Neurosci 2017; 40: 189–210, https://doi.org/10.1146/annurev-neuro-072116-031153.
- Hu J., Sha W., Yuan S., Wu J., Huang Y. Aggregation, transmission, and toxicity of the microtubule-associated protein tau: a complex comprehension. Int J Mol Sci 2023; 24(19): 15023, https://doi.org/10.3390/ijms241915023.
- Alonso A.D.C., El Idrissi A., Candia R., Morozova V., Kleiman F.E. Tau: more than a microtubule-binding protein in neurons. Cytoskeleton (Hoboken) 2024; 81(1): 71–77, https://doi.org/10.1002/cm.21795.
- Boyarko B., Hook V. Human Tau isoforms and proteolysis for production of toxic tau fragments in neurodegeneration. Front Neurosci 2021; 15: 702788, https://doi.org/10.3389/fnins.2021.702788.
- Rösler T.W., Tayaranian Marvian A., Brendel M., Nykänen N.P., Höllerhage M., Schwarz S.C., Hopfner F., Koeglsperger T., Respondek G., Schweyer K., Levin J., Villemagne V.L., Barthel H., Sabri O., Müller U., Meissner W.G., Kovacs G.G., Höglinger G.U. Four-repeat tauopathies. Prog Neurobiol 2019; 180: 101644, https://doi.org/10.1016/j.pneurobio.2019.101644.
- Chaudhry A., Houlden H., Rizig M. Novel fluid biomarkers to differentiate frontotemporal dementia and dementia with Lewy bodies from Alzheimer’s disease: a systematic review. J Neurol Sci 2020; 415: 116886, https://doi.org/10.1016/j.jns.2020.116886.
- Barthélemy N.R., Liu H., Lu W., Kotzbauer P.T., Bateman R.J., Lucey B.P. Sleep deprivation affects tau phosphorylation in human cerebrospinal fluid. Ann Neurol 2020; 87(5): 700–709, https://doi.org/10.1002/ana.25702.
- Leuzy A., Janelidze S., Mattsson-Carlgren N., Palmqvist S., Jacobs D., Cicognola C., Stomrud E., Vanmechelen E., Dage J.L., Hansson O. Comparing the clinical utility and diagnostic performance of CSF P-Tau181, P-Tau217, and P-Tau231 assays. Neurology 2021; 97(17): e1681–e1694, https://doi.org/10.1212/WNL.0000000000012727.
- Therriault J., Vermeiren M., Servaes S., Tissot C., Ashton N.J., Benedet A.L., Karikari T.K., Lantero-Rodriguez J., Brum W.S., Lussier F.Z., Bezgin G., Stevenson J., Rahmouni N., Kunach P., Wang Y.T., Fernandez-Arias J., Socualaya K.Q., Macedo A.C., Ferrari-Souza J.P., Ferreira P.C.L., Bellaver B., Leffa D.T., Zimmer E.R., Vitali P., Soucy J.P., Triana-Baltzer G., Kolb H.C., Pascoal T.A., Saha-Chaudhuri P., Gauthier S., Zetterberg H., Blennow K., Rosa-Neto P. Association of phosphorylated tau biomarkers with amyloid positron emission tomography vs tau positron emission tomography. JAMA Neurol 2023; 80(2): 188–199, https://doi.org/10.1001/jamaneurol.2022.4485.
- Carnazza K.E., Komer L.E., Xie Y.X., Pineda A., Briano J.A., Gao V., Na Y., Ramlall T., Buchman V.L., Eliezer D., Sharma M., Burré J. Synaptic vesicle binding of α-synuclein is modulated by β- and γ-synucleins. Cell Rep 2022; 39(2): 110675, https://doi.org/10.1016/j.celrep.2022.110675.
- Calabresi P., Mechelli A., Natale G., Volpicelli-Daley L., Di Lazzaro G., Ghiglieri V. Alpha-synuclein in Parkinson’s disease and other synucleinopathies: from overt neurodegeneration back to early synaptic dysfunction. Cell Death Dis 2023; 14(3): 176, https://doi.org/10.1038/s41419-023-05672-9.
- Yang Y., Shi Y., Schweighauser M., Zhang X., Kotecha A., Murzin A.G., Garringer H.J., Cullinane P.W., Saito Y., Foroud T., Warner T.T., Hasegawa K., Vidal R., Murayama S., Revesz T., Ghetti B., Hasegawa M., Lashley T., Scheres S.H.W., Goedert M. Structures of α-synuclein filaments from human brains with Lewy pathology. Nature 2022; 610(7933): 791–795, https://doi.org/10.1038/s41586-022-05319-3.
- Estaun-Panzano J., Arotcarena M.L., Bezard E. Monitoring α-synuclein aggregation. Neurobiol Dis 2023; 176: 105966, https://doi.org/10.1016/j.nbd.2022.105966.
- Dovidchenko N.V., Leonova E.I., Galzitskaya O.V. Mechanisms of amyloid fibril formation. Biochemistry (Mosc) 2014; 79(13): 1515–1527, https://doi.org/10.1134/S0006297914130057.
- Kachkin D.V., Volkov K.V., Sopova J.V., Bobylev A.G., Fedotov S.A., Inge-Vechtomov S.G., Galzitskaya O.V., Chernoff Y.O., Rubel A.A., Aksenova A.Y. Human RAD51 protein forms amyloid-like aggregates in vitro. Int J Mol Sci 2022; 23(19): 11657, https://doi.org/10.3390/ijms231911657.
- Fitzpatrick A.W., Debelouchina G.T., Bayro M.J., Clare D.K., Caporini M.A., Bajaj V.S., Jaroniec C.P., Wang L., Ladizhansky V., Müller S.A., MacPhee C.E., Waudby C.A., Mott H.R., De Simone A., Knowles T.P., Saibil H.R., Vendruscolo M., Orlova E.V., Griffin R.G., Dobson C.M. Atomic structure and hierarchical assembly of a cross-β amyloid fibril. Proc Natl Acad Sci U S A 2013; 110(14): 5468–5473, https://doi.org/10.1073/pnas.1219476110.
- Min J.H., Sarlus H., Oasa S., Harris R.A. Thioflavin-T: application as a neuronal body and nucleolar stain and the blue light photo enhancement effect. Sci Rep 2024; 14(1): 24846, https://doi.org/10.1038/s41598-024-74359-8.
- Goldy J.N., Youker R.T. Characterization of spatial differences in two misfolded proteins during aggresome formation. MicroPubl Biol 2024, https://doi.org/10.17912/micropub.biology.001312.
- Kaur G., Poljak A., Braidy N., Crawford J.D., Lo J., Sachdev P.S. Fluid biomarkers and APOE status of early onset Alzheimer’s disease variants: a systematic review and meta-analysis. J Alzheimers Dis 2020; 75(3): 827–843, https://doi.org/10.3233/JAD-200052.
- Belloy M.E., Andrews S.J., Le Guen Y., Cuccaro M., Farrer L.A., Napolioni V., Greicius M.D. APOE genotype and Alzheimer disease risk across age, sex, and population ancestry. JAMA Neurol 2023; 80(12): 1284–1294, https://doi.org/10.1001/jamaneurol.2023.3599.
- Madore C., Yin Z., Leibowitz J., Butovsky O. Microglia, lifestyle stress, and neurodegeneration. Immunity 2020; 52(2): 222–240, https://doi.org/10.1016/j.immuni.2019.12.003.
- Bantle C.M., Phillips A.T., Smeyne R.J., Rocha S.M., Olson K.E., Tjalkens R.B. Infection with mosquito-borne alphavirus induces selective loss of dopaminergic neurons, neuroinflammation and widespread protein aggregation. NPJ Parkinsons Dis 2019; 5: 20, https://doi.org/10.1038/s41531-019-0090-8.
- Han X., Sun S., Sun Y., Song Q., Zhu J., Song N., Chen M., Sun T., Xia M., Ding J., Lu M., Yao H., Hu G. Small molecule-driven NLRP3 inflammation inhibition via interplay between ubiquitination and autophagy: implications for Parkinson disease. Autophagy 2019; 15(11): 1860–1881, https://doi.org/10.1080/15548627.2019.1596481.
- Won S.J., Fong R., Butler N., Sanchez J., Zhang Y., Wong C., Tambou Nzoutchoum O., Huynh A., Pan J., Swanson R.A. Neuronal oxidative stress promotes α-synuclein aggregation in vivo. Antioxidants (Basel) 2022; 11(12): 2466, https://doi.org/10.3390/antiox11122466.
- Wegmann S., Biernat J., Mandelkow E. A current view on tau protein phosphorylation in Alzheimer’s disease. Curr Opin Neurobiol 2021; 69: 131–138, https://doi.org/10.1016/j.conb.2021.03.003.
- Conway K.A., Lee S.J., Rochet J.C., Ding T.T., Williamson R.E., Lansbury P.T. Jr. Acceleration of oligomerization, not fibrillization, is a shared property of both alpha-synuclein mutations linked to early-onset Parkinson’s disease: implications for pathogenesis and therapy. Proc Natl Acad Sci U S A 2000; 97(2): 571–576, https://doi.org/10.1073/pnas.97.2.571.
- Sgarbossa A. Natural biomolecules and protein aggregation: emerging strategies against amyloidogenesis. Int J Mol Sci 2012; 13(12): 17121–17137, https://doi.org/10.3390/ijms131217121.
- Harsolia R.S., Kanwar A., Gour S., Kumar V., Kumar V., Bansal R., Kumar S., Singh M., Yadav J.K. Predicted aggregation-prone region (APR) in βB1-crystallin forms the amyloid-like structure and induces aggregation of soluble proteins isolated from human cataractous eye lens. Int J Biol Macromol 2020; 163: 702–710, https://doi.org/10.1016/j.ijbiomac.2020.07.028.
- Housmans J.A.J., Wu G., Schymkowitz J., Rousseau F. A guide to studying protein aggregation. FEBS J 2023; 290(3): 554–583, https://doi.org/10.1111/febs.16312.
- Braak H., Del Tredici K., Rüb U., de Vos R.A., Jansen Steur E.N., Braak E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol Aging 2003; 24(2): 197–211, https://doi.org/10.1016/s0197-4580(02)00065-9.
- Horsager J., Knudsen K., Sommerauer M. Clinical and imaging evidence of brain-first and body-first Parkinson’s disease. Neurobiol Dis 2022; 164: 105626, https://doi.org/10.1016/j.nbd.2022.105626.
- Braak H., Braak E. Neuropathological stageing of Alzheimer-related changes. Acta Neuropathol 1991; 82(4): 239–259, https://doi.org/10.1007/BF00308809.
- Trejo-Lopez J.A., Yachnis A.T., Prokop S. Neuropathology of Alzheimer’s disease. Neurotherapeutics 2022; 19(1): 173–185, https://doi.org/10.1007/s13311-021-01146-y.
- Macedo A.C., Tissot C., Therriault J., Servaes S., Wang Y.T., Fernandez-Arias J., Rahmouni N., Lussier F.Z., Vermeiren M., Bezgin G., Vitali P., Ng K.P., Zimmer E.R., Guiot M.C., Pascoal T.A., Gauthier S., Rosa-Neto P. The use of tau PET to stage Alzheimer disease according to the braak staging framework. J Nucl Med 2023; 64(8): 1171–1178, https://doi.org/10.2967/jnumed.122.265200.
- Leng K., Li E., Eser R., Piergies A., Sit R., Tan M., Neff N., Li S.H., Rodriguez R.D., Suemoto C.K., Leite R.E.P., Ehrenberg A.J., Pasqualucci C.A., Seeley W.W., Spina S., Heinsen H., Grinberg L.T., Kampmann M. Molecular characterization of selectively vulnerable neurons in Alzheimer’s disease. Nat Neurosci 2021; 24(2): 276–287, https://doi.org/10.1038/s41593-020-00764-7.
- Yang S., Park J.H., Lu H.C. Axonal energy metabolism, and the effects in aging and neurodegenerative diseases. Mol Neurodegener 2023; 18(1): 49, https://doi.org/10.1186/s13024-023-00634-3.
- Zimmer T.S., Orr A.L., Orr A.G. Astrocytes in selective vulnerability to neurodegenerative disease. Trends Neurosci 2024; 47(4): 289–302, https://doi.org/10.1016/j.tins.2024.02.008.
- Ma J., Gao J., Wang J., Xie A. Prion-like mechanisms in Parkinson’s disease. Front Neurosci 2019; 13: 552, https://doi.org/10.3389/fnins.2019.00552.
- da Silva Correia A., Schmitz M., Fischer A.L., da Silva Correia S., Simonetti F.L., Saher G., Goya-Maldonado R., Arora A.S., Fischer A., Outeiro T.F., Zerr I. Cellular prion protein acts as mediator of amyloid beta uptake by caveolin-1 causing cellular dysfunctions in vitro and in vivo. Alzheimers Dement 2024; 20(10): 6776–6792, https://doi.org/10.1002/alz.14120.
- Renner M., Melki R. Protein aggregation and prionopathies. Pathol Biol (Paris) 2014; 62(3): 162–168, https://doi.org/10.1016/j.patbio.2014.01.003.
- Condello C., Westaway D., Prusiner S.B. Expanding the prion paradigm to include Alzheimer and Parkinson diseases. JAMA Neurol 2024; 81(10): 1023–1024, https://doi.org/10.1001/jamaneurol.2024.2464.
- Aulić S., Masperone L., Narkiewicz J., Isopi E., Bistaffa E., Ambrosetti E., Pastore B., De Cecco E., Scaini D., Zago P., Moda F., Tagliavini F., Legname G. α-Synuclein amyloids hijack prion protein to gain cell entry, facilitate cell-to-cell spreading and block prion replication. Sci Rep 2017; 7(1): 10050, https://doi.org/10.1038/s41598-017-10236-x.
- Brás I.C., Lopes L.V., Outeiro T.F. Sensing α-synuclein from the outside via the prion protein: implications for neurodegeneration. Mov Disord 2018; 33(11): 1675–1684, https://doi.org/10.1002/mds.27478.
- Clavaguera F., Hench J., Goedert M., Tolnay M. Invited review: prion-like transmission and spreading of tau pathology. Neuropathol Appl Neurobiol 2015; 41(1): 47–58, https://doi.org/10.1111/nan.12197.
- Volpicelli-Daley L.A., Luk K.C., Lee V.M. Addition of exogenous α-synuclein preformed fibrils to primary neuronal cultures to seed recruitment of endogenous α-synuclein to Lewy body and Lewy neurite-like aggregates. Nat Protoc 2014; 9(9): 2135–2146, https://doi.org/10.1038/nprot.2014.143.
- Tarutani A., Suzuki G., Shimozawa A., Nonaka T., Akiyama H., Hisanaga S., Hasegawa M. The effect of fragmented pathogenic α-synuclein seeds on prion-like propagation. J Biol Chem 2016; 291(36): 18675–18688, https://doi.org/10.1074/jbc.M116.734707.
- Novotny R., Langer F., Mahler J., Skodras A., Vlachos A., Wegenast-Braun B.M., Kaeser S.A., Neher J.J., Eisele Y.S., Pietrowski M.J., Nilsson K.P., Deller T., Staufenbiel M., Heimrich B., Jucker M. Conversion of synthetic Aβ to in vivo active seeds and amyloid plaque formation in a hippocampal slice culture model. J Neurosci 2016; 36(18): 5084–5093, https://doi.org/10.1523/JNEUROSCI.0258-16.2016.
- Kane M.D., Lipinski W.J., Callahan M.J., Bian F., Durham R.A., Schwarz R.D., Roher A.E., Walker L.C. Evidence for seeding of beta-amyloid by intracerebral infusion of Alzheimer brain extracts in beta-amyloid precursor protein-transgenic mice. J Neurosci 2000; 20(10): 3606–3611, https://doi.org/10.1523/JNEUROSCI.20-10-03606.2000.
- Eisele Y.S., Bolmont T., Heikenwalder M., Langer F., Jacobson L.H., Yan Z.X., Roth K., Aguzzi A., Staufenbiel M., Walker L.C., Jucker M. Induction of cerebral beta-amyloidosis: intracerebral versus systemic Abeta inoculation. Proc Natl Acad Sci U S A 2009; 106(31): 12926–12931, https://doi.org/10.1073/pnas.0903200106.
- He Z., Guo J.L., McBride J.D., Narasimhan S., Kim H., Changolkar L., Zhang B., Gathagan R.J., Yue C., Dengler C., Stieber A., Nitla M., Coulter D.A., Abel T., Brunden K.R., Trojanowski J.Q., Lee V.M. Amyloid-β plaques enhance Alzheimer’s brain tau-seeded pathologies by facilitating neuritic plaque tau aggregation. Nat Med 2018; 24(1): 29–38, https://doi.org/10.1038/nm.4443.
- Li X., Ospitalieri S., Robberechts T., Hofmann L., Schmid C., Rijal Upadhaya A., Koper M.J., von Arnim C.A.F., Kumar S., Willem M., Gnoth K., Ramakers M., Schymkowitz J., Rousseau F., Walter J., Ronisz A., Balakrishnan K., Thal D.R. Seeding, maturation and propagation of amyloid β-peptide aggregates in Alzheimer’s disease. Brain 2022; 145(10): 3558–3570, https://doi.org/10.1093/brain/awac202.
- Gary C., Lam S., Hérard A.S., Koch J.E., Petit F., Gipchtein P., Sawiak S.J., Caillierez R., Eddarkaoui S., Colin M., Aujard F., Deslys J.P.; French Neuropathology Network; Brouillet E., Buée L., Comoy E.E., Pifferi F., Picq J.L., Dhenain M. Encephalopathy induced by Alzheimer brain inoculation in a non-human primate. Acta Neuropathol Commun 2019; 7(1): 126, https://doi.org/10.1186/s40478-019-0771-x.
- Boi L., Pisanu A., Palmas M.F., Fusco G., Carboni E., Casu M.A., Satta V., Scherma M., Janda E., Mocci I., Mulas G., Ena A., Spiga S., Fadda P., De Simone A., Carta A.R. Modeling Parkinson’s disease neuropathology and symptoms by intranigral inoculation of preformed human α-synuclein oligomers. Int J Mol Sci 2020; 21(22): 8535, https://doi.org/10.3390/ijms21228535.
- Okuda S., Nakayama T., Uemura N., Hikawa R., Ikuno M., Yamakado H., Inoue H., Tachibana N., Hayashi Y., Takahashi R., Egawa N. Striatal-inoculation of α-synuclein preformed fibrils aggravated the phenotypes of REM sleep without atonia in A53T BAC-SNCA transgenic mice. Int J Mol Sci 2022; 23(21): 13390, https://doi.org/10.3390/ijms232113390.
- Flores-Cuadrado A., Saiz-Sanchez D., Mohedano-Moriano A., Martinez-Marcos A., Ubeda-Bañon I. Neurodegeneration and contralateral α-synuclein induction after intracerebral α-synuclein injections in the anterior olfactory nucleus of a Parkinson’s disease A53T mouse model. Acta Neuropathol Commun 2019; 7(1): 56, https://doi.org/10.1186/s40478-019-0713-7.
- Brackhan M., Calza G., Lundgren K., Bascuñana P., Brüning T., Soliymani R., Kumar R., Abelein A., Baumann M., Lalowski M., Pahnke J. Isotope-labeled amyloid-β does not transmit to the brain in a prion-like manner after peripheral administration. EMBO Rep 2022; 23(7): e54405, https://doi.org/10.15252/embr.202154405.
- Challis C., Hori A., Sampson T.R., Yoo B.B., Challis R.C., Hamilton A.M., Mazmanian S.K., Volpicelli-Daley L.A., Gradinaru V. Gut-seeded α-synuclein fibrils promote gut dysfunction and brain pathology specifically in aged mice. Nat Neurosci 2020; 23(3): 327–336, https://doi.org/10.1038/s41593-020-0589-7.
- Ferrer I., Zelaya M.V., Aguiló García M., Carmona M., López-González I., Andrés-Benito P., Lidón L., Gavín R., Garcia-Esparcia P., Del Rio J.A. Relevance of host tau in tau seeding and spreading in tauopathies. Brain Pathol 2020; 30(2): 298–318, https://doi.org/10.1111/bpa.12778.
- Ferrer I., Andrés-Benito P., Garcia-Esparcia P., López-Gonzalez I., Valiente D., Jordán-Pirla M., Carmona M., Sala-Jarque J., Gil V., Del Rio J.A. Differences in tau seeding in newborn and adult wild-type mice. Int J Mol Sci 2022; 23(9): 4789, https://doi.org/10.3390/ijms23094789.
- Williams T., Sorrentino Z., Weinrich M., Giasson B.I., Chakrabarty P. Differential cross-seeding properties of tau and α-synuclein in mouse models of tauopathy and synucleinopathy. Brain Commun 2020; 2(2): fcaa090, https://doi.org/10.1093/braincomms/fcaa090.
- Pan L., Li C., Meng L., Tian Y., He M., Yuan X., Zhang G., Zhang Z., Xiong J., Chen G., Zhang Z. Tau accelerates α-synuclein aggregation and spreading in Parkinson’s disease. Brain 2022; 145(10): 3454–3471, https://doi.org/10.1093/brain/awac171.
- Bassil F., Brown H.J., Pattabhiraman S., Iwasyk J.E., Maghames C.M., Meymand E.S., Cox T.O., Riddle D.M., Zhang B., Trojanowski J.Q., Lee V.M. Amyloid-beta (Aβ) plaques promote seeding and spreading of alpha-synuclein and tau in a mouse model of Lewy body disorders with Aβ pathology. Neuron 2020; 105(2): 260–275.e6, https://doi.org/10.1016/j.neuron.2019.10.010.
- Hamaguchi T., Taniguchi Y., Sakai K., Kitamoto T., Takao M., Murayama S., Iwasaki Y., Yoshida M., Shimizu H., Kakita A., Takahashi H., Suzuki H., Naiki H., Sanjo N., Mizusawa H., Yamada M. Significant association of cadaveric dura mater grafting with subpial Aβ deposition and meningeal amyloid angiopathy. Acta Neuropathol 2016; 132(2): 313–315, https://doi.org/10.1007/s00401-016-1588-3.
- Peng C., Trojanowski J.Q., Lee V.M. Protein transmission in neurodegenerative disease. Nat Rev Neurol 2020; 16(4): 199–212, https://doi.org/10.1038/s41582-020-0333-7.
- Purro S.A., Farrow M.A., Linehan J., Nazari T., Thomas D.X., Chen Z., Mengel D., Saito T., Saido T., Rudge P., Brandner S., Walsh D.M., Collinge J. Transmission of amyloid-β protein pathology from cadaveric pituitary growth hormone. Nature 2018; 564(7736): 415–419, https://doi.org/10.1038/s41586-018-0790-y.
- Banerjee G., Farmer S.F., Hyare H., Jaunmuktane Z., Mead S., Ryan N.S., Schott J.M., Werring D.J., Rudge P., Collinge J. Iatrogenic Alzheimer’s disease in recipients of cadaveric pituitary-derived growth hormone. Nat Med 2024; 30(2): 394–402, https://doi.org/10.1038/s41591-023-02729-2.
- Tong M.K., Thakur A., Yang T., Wong S.K., Li W.K., Lee Y. Amyloid-β oligomer-induced neurotoxicity by exosomal interactions between neuron and microglia. Biochem Biophys Res Commun 2024; 727: 150312, https://doi.org/10.1016/j.bbrc.2024.150312.
- Ngolab J., Trinh I., Rockenstein E., Mante M., Florio J., Trejo M., Masliah D., Adame A., Masliah E., Rissman R.A. Brain-derived exosomes from dementia with Lewy bodies propagate α-synuclein pathology. Acta Neuropathol Commun 2017; 5(1): 46, https://doi.org/10.1186/s40478-017-0445-5.
- Melachroinou K., Divolis G., Tsafaras G., Karampetsou M., Fortis S., Stratoulias Y., Papadopoulou G., Kriebardis A.G., Samiotaki M., Vekrellis K. Endogenous alpha-synuclein is essential for the transfer of pathology by exosome-enriched extracellular vesicles, following inoculation with preformed fibrils in vivo. Aging Dis 2024; 15(2): 869–892, https://doi.org/10.14336/AD.2023.0614.
- Guo M., Wang J., Zhao Y., Feng Y., Han S., Dong Q., Cui M., Tieu K. Microglial exosomes facilitate α-synuclein transmission in Parkinson’s disease. Brain 2020; 143(5): 1476–1497, https://doi.org/10.1093/brain/awaa090.
- Polanco J.C., Hand G.R., Briner A., Li C., Götz J. Exosomes induce endolysosomal permeabilization as a gateway by which exosomal tau seeds escape into the cytosol. Acta Neuropathol 2021; 141(2): 235–256, https://doi.org/10.1007/s00401-020-02254-3.
- Jain N., Ulrich J.D. TREM2 and microglia exosomes: a potential highway for pathological tau. Mol Neurodegener 2022; 17(1): 73, https://doi.org/10.1186/s13024-022-00581-5.
- Sardar Sinha M., Ansell-Schultz A., Civitelli L., Hildesjö C., Larsson M., Lannfelt L., Ingelsson M., Hallbeck M. Alzheimer’s disease pathology propagation by exosomes containing toxic amyloid-beta oligomers. Acta Neuropathol 2018; 136(1): 41–56, https://doi.org/10.1007/s00401-018-1868-1.
- Ruan Z., Pathak D., Venkatesan Kalavai S., Yoshii-Kitahara A., Muraoka S., Bhatt N., Takamatsu-Yukawa K., Hu J., Wang Y., Hersh S., Ericsson M., Gorantla S., Gendelman H.E., Kayed R., Ikezu S., Luebke J.I., Ikezu T. Alzheimer’s disease brain-derived extracellular vesicles spread tau pathology in interneurons. Brain 2021; 144(1): 288–309, https://doi.org/10.1093/brain/awaa376.
- Wei S., Ma X., Chen Y., Wang J., Hu L., Liu Z., Mo L., Zhou N., Chen W., Zhu H., Yan S. Alzheimer’s disease-derived outer membrane vesicles exacerbate cognitive dysfunction, modulate the gut microbiome, and increase neuroinflammation and amyloid-β production. Mol Neurobiol 2025; 62(4): 5109–5132, https://doi.org/10.1007/s12035-024-04579-6.
- Liu W.L., Lin H.W., Lin M.R., Yu Y., Liu H.H., Dai Y.L., Chen L.W., Jia W.W., He X.J., Li X.L., Zhu J.F., Xue X.H., Tao J., Chen L.D. Emerging blood exosome-based biomarkers for preclinical and clinical Alzheimer’s disease: a meta-analysis and systematic review. Neural Regen Res 2022; 17(11): 2381–2390, https://doi.org/10.4103/1673-5374.335832.
- Kluge A., Bunk J., Schaeffer E., Drobny A., Xiang W., Knacke H., Bub S., Lückstädt W., Arnold P., Lucius R., Berg D., Zunke F. Detection of neuron-derived pathological α-synuclein in blood. Brain 2022; 145(9): 3058–3071, https://doi.org/10.1093/brain/awac115.
- Kluge A., Schaeffer E., Bunk J., Sommerauer M., Röttgen S., Schulte C., Roeben B., von Thaler A.-K., Welzel J., Lucius R., Heinzel S., Xiang W., Eschweiler G.W., Maetzler W., Suenkel U., Berg D. Detecting misfolded α-synuclein in blood years before the diagnosis of Parkinson’s disease. Mov Disord 2024; 39: 1289–1299, https://doi.org/10.1002/mds.29766.
- Mothes T., Portal B., Konstantinidis E., Eltom K., Libard S., Streubel-Gallasch L., Ingelsson M., Rostami J., Lindskog M., Erlandsson A. Astrocytic uptake of neuronal corpses promotes cell-to-cell spreading of tau pathology. Acta Neuropathol Commun 2023; 11(1): 97, https://doi.org/10.1186/s40478-023-01589-8.
- Rajasekaran S., Witt S.N. Trojan horses and tunneling nanotubes enable α-synuclein pathology to spread in Parkinson disease. PLoS Biol 2021; 19(7): e3001331, https://doi.org/10.1371/journal.pbio.3001331.
- Scheiblich H., Eikens F., Wischhof L., Opitz S., Jüngling K., Cserép C., Schmidt S.V., Lambertz J., Bellande T., Pósfai B., Geck C., Spitzer J., Odainic A., Castro-Gomez S., Schwartz S., Boussaad I., Krüger R., Glaab E., Di Monte D.A., Bano D., Dénes Á., Latz E., Melki R., Pape H.C., Heneka M.T. Microglia rescue neurons from aggregate-induced neuronal dysfunction and death through tunneling nanotubes. Neuron 2024; 112(18): 3106–3125.e8, https://doi.org/10.1016/j.neuron.2024.06.029.
- Chakraborty R., Nonaka T., Hasegawa M., Zurzolo C. Tunnelling nanotubes between neuronal and microglial cells allow bi-directional transfer of α-synuclein and mitochondria. Cell Death Dis 2023; 14(5): 329, https://doi.org/10.1038/s41419-023-05835-8.
- Xu Y., Du S., Marsh J.A., Horie K., Sato C., Ballabio A., Karch C.M., Holtzman D.M., Zheng H. TFEB regulates lysosomal exocytosis of tau and its loss of function exacerbates tau pathology and spreading. Mol Psychiatry 2021; 26(10): 5925–5939, https://doi.org/10.1038/s41380-020-0738-0.
- Shearer L.J., Petersen N.O., Woodside M.T. Internalization of α-synuclein oligomers into SH-SY5Y cells. Biophys J 2021; 120(5): 877–885, https://doi.org/10.1016/j.bpj.2020.12.031.
- Zhang S., Liu Y.Q., Jia C., Lim Y.J., Feng G., Xu E., Long H., Kimura Y., Tao Y., Zhao C., Wang C., Liu Z., Hu J.J., Ma M.R., Liu Z., Jiang L., Li D., Wang R., Dawson V.L., Dawson T.M., Li Y.M., Mao X., Liu C. Mechanistic basis for receptor-mediated pathological α-synuclein fibril cell-to-cell transmission in Parkinson’s disease. Proc Natl Acad Sci U S A 2021; 118(26): e2011196118, https://doi.org/10.1073/pnas.2011196118.
- Shi J.M., Zhu L., Lan X., Zhao D.W., He Y.J., Sun Z.Q., Wu D., Li H.Y. Endocytosis is a key mode of interaction between extracellular β-amyloid and the cell membrane. Biophys J 2020; 119(6): 1078–1090, https://doi.org/10.1016/j.bpj.2020.07.035.
- Lungu O., Bares M. Editorial: neuropsychology through the MRI looking glass. Front Neurol 2020; 11: 609897, https://doi.org/10.3389/fneur.2020.609897.
- Bousiges O., Blanc F. Biomarkers of dementia with Lewy bodies: differential diagnostic with Alzheimer’s disease. Int J Mol Sci 2022; 23(12): 6371, https://doi.org/10.3390/ijms23126371.
- Smailagic N., Vacante M., Hyde C., Martin S., Ukoumunne O., Sachpekidis C. 18F-FDG PET for the early diagnosis of Alzheimer’s disease dementia and other dementias in people with mild cognitive impairment (MCI). Cochrane Database Syst Rev 2015; 1(1): CD010632, https://doi.org/10.1002/14651858.CD010632.pub2.
- Lombardi G., Crescioli G., Cavedo E., Lucenteforte E., Casazza G., Bellatorre A.G., Lista C., Costantino G., Frisoni G., Virgili G., Filippini G. Structural magnetic resonance imaging for the early diagnosis of dementia due to Alzheimer’s disease in people with mild cognitive impairment. Cochrane Database Syst Rev 2020; 3(3): CD009628, https://doi.org/10.1002/14651858.CD009628.pub2.
- Solje E., Benussi A., Buratti E., Remes A.M., Haapasalo A., Borroni B. State-of-the-art methods and emerging fluid biomarkers in the diagnostics of dementia — a short review and diagnostic algorithm. Diagnostics (Basel) 2021; 11(5): 788, https://doi.org/10.3390/diagnostics11050788.
- Skillbäck T., Farahmand B.Y., Rosén C., Mattsson N., Nägga K., Kilander L., Religa D., Wimo A., Winblad B., Schott J.M., Blennow K., Eriksdotter M., Zetterberg H. Cerebrospinal fluid tau and amyloid-β1-42 in patients with dementia. Brain 2015; 138(Pt 9): 2716–2731, https://doi.org/10.1093/brain/awv181.
- Arranz J., Zhu N., Rubio-Guerra S., Rodríguez-Baz Í., Ferrer R., Carmona-Iragui M., Barroeta I., Illán-Gala I., Santos-Santos M., Fortea J., Lleó A., Tondo M., Alcolea D. Diagnostic performance of plasma pTau217, pTau181, Aβ1-42 and Aβ1-40 in the LUMIPULSE automated platform for the detection of Alzheimer disease. Alzheimers Res Ther 2024; 16(1): 139, https://doi.org/10.1186/s13195-024-01513-9.
- Gąsior-Głogowska M.E., Szulc N., Szefczyk M. Challenges in experimental methods. Methods Mol Biol 2022; 2340: 281–307, https://doi.org/10.1007/978-1-0716-1546-1_13.
- Guerrero-Ferreira R., Taylor N.M., Arteni A.A., Kumari P., Mona D., Ringler P., Britschgi M., Lauer M.E., Makky A., Verasdonck J., Riek R., Melki R., Meier B.H., Böckmann A., Bousset L., Stahlberg H. Two new polymorphic structures of human full-length alpha-synuclein fibrils solved by cryo-electron microscopy. Elife 2019; 8: e48907, https://doi.org/10.7554/eLife.48907.
- Motta C., Di Donna M.G., Bonomi C.G., Assogna M., Chiaravalloti A., Mercuri N.B., Koch G., Martorana A. Different associations between amyloid-βeta 42, amyloid-βeta 40, and amyloid-βeta 42/40 with soluble phosphorylated-tau and disease burden in Alzheimer’s disease: a cerebrospinal fluid and fluorodeoxyglucose-positron emission tomography study. Alzheimers Res Ther 2023; 15(1): 144, https://doi.org/10.1186/s13195-023-01291-w.
- Santos J.R.F., Bauer C., Schuchhardt J., Wedekind D., Waniek K., Lachmann I., Wiltfang J., Vogelgsang J. Validation of a prototype tau Thr231 phosphorylation CSF ELISA as a potential biomarker for Alzheimer’s disease. J Neural Transm (Vienna) 2019; 126(3): 339–348, https://doi.org/10.1007/s00702-019-01982-5.
- Kovalenko Ye.A., Makhnovich Ye.V., Pervunina A.V., Akimov K.A., Marahovskaya Ye.A., Bogolepova A.N. Biomarkers of blood in the early diagnosis of Alzheimer’s disease. Effektivnaya farmakoterapiya 2023; 19(45): 30–36.
- Shinohara M., Hirokawa J., Shimodaira A., Tashiro Y., Suzuki K., Gheni G., Fukumori A., Matsubara T., Morishima M., Saito Y., Murayama S., Sato N. ELISA evaluation of tau accumulation in the brains of patients with Alzheimer disease. J Neuropathol Exp Neurol 2021; 80(7): 652–662, https://doi.org/10.1093/jnen/nlab047.
- Herrmann M., Golombowski S., Kräuchi K., Frey P., Mourton-Gilles C., Hulette C., Rosenberg C., Müller-Spahn F., Hock C. ELISA-quantitation of phosphorylated tau protein in the Alzheimer’s disease brain. Eur Neurol 1999; 42(4): 205–210, https://doi.org/10.1159/000008108.
- Teunissen C.E., Kimble L., Bayoumy S., Bolsewig K., Burtscher F., Coppens S., Das S., Gogishvili D., Fernandes Gomes B., Gómez de San José N., Mavrina E., Meda F.J., Mohaupt P., Mravinacová S., Waury K., Wojdała A.L., Abeln S., Chiasserini D., Hirtz C., Gaetani L., Vermunt L., Bellomo G., Halbgebauer S., Lehmann S., Månberg A., Nilsson P., Otto M., Vanmechelen E., Verberk I.M.W., Willemse E., Zetterberg H.; MIRIADE consortium. Methods to discover and validate biofluid-based biomarkers in neurodegenerative dementias. Mol Cell Proteomics 2023; 22(10): 100629, https://doi.org/10.1016/j.mcpro.2023.100629.
- Truffi M., Garofalo M., Ricciardi A., Cotta Ramusino M., Perini G., Scaranzin S., Gastaldi M., Albasini S., Costa A., Chiavetta V., Corsi F., Morasso C., Gagliardi S. Neurofilament-light chain quantification by Simoa and Ella in plasma from patients with dementia: a comparative study. Sci Rep 2023; 13(1): 4041, https://doi.org/10.1038/s41598-023-29704-8.
- Carrette O., Demalte I., Scherl A., Yalkinoglu O., Corthals G., Burkhard P., Hochstrasser D.F., Sanchez J.C. A panel of cerebrospinal fluid potential biomarkers for the diagnosis of Alzheimer’s disease. Proteomics 2003; 3(8): 1486–1494, https://doi.org/10.1002/pmic.200300470.
- Conrotto P., Souchelnytskyi S. Proteomic approaches in biological and medical sciences: principles and applications. Exp Oncol 2008; 30(3): 171–180.
- Bader J.M., Geyer P.E., Müller J.B., Strauss M.T., Koch M., Leypoldt F., Koertvelyessy P., Bittner D., Schipke C.G., Incesoy E.I., Peters O., Deigendesch N., Simons M., Jensen M.K., Zetterberg H., Mann M. Proteome profiling in cerebrospinal fluid reveals novel biomarkers of Alzheimer’s disease. Mol Syst Biol 2020; 16(6): e9356, https://doi.org/10.15252/msb.20199356.
- Karayel O., Virreira Winter S., Padmanabhan S., Kuras Y.I., Vu D.T., Tuncali I., Merchant K., Wills A.M., Scherzer C.R., Mann M. Proteome profiling of cerebrospinal fluid reveals biomarker candidates for Parkinson’s disease. Cell Rep Med 2022; 3(6): 100661, https://doi.org/10.1016/j.xcrm.2022.100661.
- Teunissen C.E., Elias N., Koel-Simmelink M.J., Durieux-Lu S., Malekzadeh A., Pham T.V., Piersma S.R., Beccari T., Meeter L.H., Dopper E.G., van Swieten J.C., Jimenez C.R., Pijnenburg Y.A. Novel diagnostic cerebrospinal fluid biomarkers for pathologic subtypes of frontotemporal dementia identified by proteomics. Alzheimers Dement (Amst) 2016; 2: 86–94, https://doi.org/10.1016/j.dadm.2015.12.004.
- Laszlo Z.I., Hindley N., Sanchez Avila A., Kline R.A., Eaton S.L., Lamont D.J., Smith C., Spires-Jones T.L., Wishart T.M., Henstridge C.M. Synaptic proteomics reveal distinct molecular signatures of cognitive change and C9ORF72 repeat expansion in the human ALS cortex. Acta Neuropathol Commun 2022; 10(1): 156, https://doi.org/10.1186/s40478-022-01455-z.
- Flynn J.D., Lee J.C. Raman fingerprints of amyloid structures. Chem Commun (Camb) 2018; 54(51): 6983–6986, https://doi.org/10.1039/c8cc03217c.
- Chen C., Qi J., Li Y., Li D., Wu L., Li R., Chen Q., Sun N. Applications of Raman spectroscopy in the diagnosis and monitoring of neurodegenerative diseases. Front Neurosci 2024; 18: 1301107, https://doi.org/10.3389/fnins.2024.1301107.
- Lochocki B., Morrema T.H.J., Ariese F., Hoozemans J.J.M., de Boer J.F. The search for a unique Raman signature of amyloid-beta plaques in human brain tissue from Alzheimer’s disease patients. Analyst 2020; 145(5): 1724–1736, https://doi.org/10.1039/c9an02087j.
- Cennamo G., Montorio D., Morra V.B., Criscuolo C., Lanzillo R., Salvatore E., Camerlingo C., Lisitskiy M., Delfino I., Portaccio M., Lepore M. Surface-enhanced Raman spectroscopy of tears: toward a diagnostic tool for neurodegenerative disease identification. J Biomed Opt 2020; 25(8): 1–12, https://doi.org/10.1117/1.JBO.25.8.087002.
- Carlomagno C., Bertazioli D., Gualerzi A., Picciolini S., Andrico M., Rodà F., Meloni M., Banfi P.I., Verde F., Ticozzi N., Silani V., Messina E., Bedoni M. Identification of the raman salivary fingerprint of Parkinson’s disease through the spectroscopic-computational combinatory approach. Front Neurosci 2021; 15: 704963, https://doi.org/10.3389/fnins.2021.704963.
- Sun J., Shi Z., Wang L., Zhang X., Luo C., Hua J., Feng M., Chen Z., Wang M., Xu C. Construction of a microcavity-based microfluidic chip with simultaneous SERS quantification of dual biomarkers for early diagnosis of Alzheimer’s disease. Talanta 2023; 261: 124677, https://doi.org/10.1016/j.talanta.2023.124677.
- Soares Martins T., Magalhães S., Rosa I.M., Vogelgsang J., Wiltfang J., Delgadillo I., Catita J., da Cruz E Silva O.A.B., Nunes A., Henriques A.G. Potential of FTIR spectroscopy applied to exosomes for Alzheimer’s disease discrimination: a pilot study. J Alzheimers Dis 2020; 74(1): 391–405, https://doi.org/10.3233/JAD-191034.
- Soares Martins T., Ferreira M., Magalhães S., Leandro K., Almeida L.P., Vogelgsang J., Breitling B., Hansen N., Esselmann H., Wiltfang J., da Cruz E Silva O.A.B., Nunes A., Henriques A.G. FTIR spectroscopy and blood-derived extracellular vesicles duo in Alzheimer’s disease. J Alzheimers Dis 2024; 98(3): 1157–1167, https://doi.org/10.3233/JAD-231239.
- Paraskevaidi M., Morais C.L.M., Lima K.M.G., Snowden J.S., Saxon J.A., Richardson A.M.T., Jones M., Mann D.M.A., Allsop D., Martin-Hirsch P.L., Martin F.L. Differential diagnosis of Alzheimer’s disease using spectrochemical analysis of blood. Proc Natl Acad Sci U S A 2017; 114(38): E7929–E7938, https://doi.org/10.1073/pnas.1701517114.
- Benseny-Cases N., Klementieva O., Cotte M., Ferrer I., Cladera J. Microspectroscopy (μFTIR) reveals co-localization of lipid oxidation and amyloid plaques in human Alzheimer disease brains. Anal Chem 2014; 86(24): 12047–12054, https://doi.org/10.1021/ac502667b.
- Palombo F., Tamagnini F., Jeynes J.C.G., Mattana S., Swift I., Nallala J., Hancock J., Brown J.T., Randall A.D., Stone N. Detection of Aβ plaque-associated astrogliosis in Alzheimer’s disease brain by spectroscopic imaging and immunohistochemistry. Analyst 2018; 143(4): 850–857, https://doi.org/10.1039/c7an01747b.
- Röhr D., Boon B.D.C., Schuler M., Kremer K., Hoozemans J.J.M., Bouwman F.H., El-Mashtoly S.F., Nabers A., Großerueschkamp F., Rozemuller A.J.M., Gerwert K. Label-free vibrational imaging of different Aβ plaque types in Alzheimer’s disease reveals sequential events in plaque development. Acta Neuropathol Commun 2020; 8(1): 222, https://doi.org/10.1186/s40478-020-01091-5.
- Gras S.L., Waddington L.J., Goldie K.N. Transmission electron microscopy of amyloid fibrils. Methods Mol Biol 2011; 752: 197–214, https://doi.org/10.1007/978-1-60327-223-0_13.
- Kollmer M., Close W., Funk L., Rasmussen J., Bsoul A., Schierhorn A., Schmidt M., Sigurdson C.J., Jucker M., Fändrich M. Cryo-EM structure and polymorphism of Aβ amyloid fibrils purified from Alzheimer’s brain tissue. Nat Commun 2019; 10(1): 4760, https://doi.org/10.1038/s41467-019-12683-8.
- Yang Y., Arseni D., Zhang W., Huang M., Lövestam S., Schweighauser M., Kotecha A., Murzin A.G., Peak-Chew S.Y., Macdonald J., Lavenir I., Garringer H.J., Gelpi E., Newell K.L., Kovacs G.G., Vidal R., Ghetti B., Ryskeldi-Falcon B., Scheres S.H.W., Goedert M. Cryo-EM structures of amyloid-β 42 filaments from human brains. Science 2022; 375(6577): 167–172, https://doi.org/10.1126/science.abm7285.
- Zhang H., Zheng X., Kwok R.T.K., Wang J., Leung N.L.C., Shi L., Sun J.Z., Tang Z., Lam J.W.Y., Qin A., Tang B.Z. In situ monitoring of molecular aggregation using circular dichroism. Nat Commun 2018; 9(1): 4961, https://doi.org/10.1038/s41467-018-07299-3.
- Waeytens J., Turbant F., Arluison V., Raussens V., Wien F. Analysis of bacterial amyloid interaction with lipidic membrane by orientated circular dichroism and infrared spectroscopies. Methods Mol Biol 2022; 2538: 217–234, https://doi.org/10.1007/978-1-0716-2529-3_15.
- Hu H.Y., Jiang L.L., Hong J.Y. Study of protein amyloid-like aggregates by solid-state circular dichroism spectroscopy. Curr Protein Pept Sci 2017; 18(1): 100–103, https://doi.org/10.2174/1389203717666160709185323.
- Rybicka A., Longhi G., Castiglioni E., Abbate S., Dzwolak W., Babenko V., Pecul M. Thioflavin T: electronic circular dichroism and circularly polarized luminescence induced by amyloid fibrils. Chemphyschem 2016; 17(18): 2931–2937, https://doi.org/10.1002/cphc.201600235.
- Protein folding, misfolding, and disease: methods and protocols. Hill A.F., Barnham K.J., Bottomley S.P., Cappai R. (editors). Humana Press; 2011, https://doi.org/10.1007/978-1-60327-223-0.
- Concha-Marambio L., Pritzkow S., Shahnawaz M., Farris C.M., Soto C. Seed amplification assay for the detection of pathologic alpha-synuclein aggregates in cerebrospinal fluid. Nat Protoc 2023; 18(4): 1179–1196, https://doi.org/10.1038/s41596-022-00787-3.
- Standke H.G., Kraus A. Seed amplification and RT-QuIC assays to investigate protein seed structures and strains. Cell Tissue Res 2023; 392(1): 323–335, https://doi.org/10.1007/s00441-022-03595-z.
- Saborio G.P., Permanne B., Soto C. Sensitive detection of pathological prion protein by cyclic amplification of protein misfolding. Nature 2001; 411(6839): 810–813, https://doi.org/10.1038/35081095.
- Saá P., Castilla J., Soto C. Ultra-efficient replication of infectious prions by automated protein misfolding cyclic amplification. J Biol Chem 2006; 281(46): 35245–35252, https://doi.org/10.1074/jbc.M603964200.
- Wang F., Pritzkow S., Soto C. PMCA for ultrasensitive detection of prions and to study disease biology. Cell Tissue Res 2023; 392(1): 307–321, https://doi.org/10.1007/s00441-022-03727-5.
- Atarashi R., Wilham J.M., Christensen L., Hughson A.G., Moore R.A., Johnson L.M., Onwubiko H.A., Priola S.A., Caughey B. Simplified ultrasensitive prion detection by recombinant PrP conversion with shaking. Nat Methods 2008; 5(3): 211–212, https://doi.org/10.1038/nmeth0308-211.
- Atarashi R., Satoh K., Sano K., Fuse T., Yamaguchi N., Ishibashi D., Matsubara T., Nakagaki T., Yamanaka H., Shirabe S., Yamada M., Mizusawa H., Kitamoto T., Klug G., McGlade A., Collins S.J., Nishida N. Ultrasensitive human prion detection in cerebrospinal fluid by real-time quaking-induced conversion. Nat Med 2011; 17(2): 175–178, https://doi.org/10.1038/nm.2294.
- Giaccone G., Moda F. PMCA applications for prion detection in peripheral tissues of patients with variant Creutzfeldt–Jakob disease. Biomolecules 2020; 10(3): 405, https://doi.org/10.3390/biom10030405.
- Rhoads D.D., Wrona A., Foutz A., Blevins J., Glisic K., Person M., Maddox R.A., Belay E.D., Schonberger L.B., Tatsuoka C., Cohen M.L., Appleby B.S. Diagnosis of prion diseases by RT-QuIC results in improved surveillance. Neurology 2020; 95(8): e1017–e1026, https://doi.org/10.1212/WNL.0000000000010086.
- Brandel J.P., Culeux A., Grznarova K., Levavasseur E., Lamy P., Privat N., Welaratne A., Denouel A., Laplanche J.L., Haik S. Amplification techniques and diagnosis of prion diseases. Rev Neurol (Paris) 2019; 175(7–8): 458–463, https://doi.org/10.1016/j.neurol.2019.06.002.
- Groveman B.R., Orrù C.D., Hughson A.G., Raymond L.D., Zanusso G., Ghetti B., Campbell K.J., Safar J., Galasko D., Caughey B. Rapid and ultra-sensitive quantitation of disease-associated α-synuclein seeds in brain and cerebrospinal fluid by αSyn RT-QuIC. Acta Neuropathol Commun 2018; 6(1): 7, https://doi.org/10.1186/s40478-018-0508-2.
- Shahnawaz M., Mukherjee A., Pritzkow S., Mendez N., Rabadia P., Liu X., Hu B., Schmeichel A., Singer W., Wu G., Tsai A.L., Shirani H., Nilsson K.P.R., Low P.A., Soto C. Discriminating α-synuclein strains in Parkinson’s disease and multiple system atrophy. Nature 2020; 578(7794): 273–277, https://doi.org/10.1038/s41586-020-1984-7.
- Bellomo G., De Luca C.M.G., Paoletti F.P., Gaetani L., Moda F., Parnetti L. α-Synuclein seed amplification assays for diagnosing synucleinopathies: the way forward. Neurology 2022; 99(5): 195–205, https://doi.org/10.1212/WNL.0000000000200878.
- Okuzumi A., Hatano T., Matsumoto G., Nojiri S., Ueno S.I., Imamichi-Tatano Y., Kimura H., Kakuta S., Kondo A., Fukuhara T., Li Y., Funayama M., Saiki S., Taniguchi D., Tsunemi T., McIntyre D., Gérardy J.J., Mittelbronn M., Kruger R., Uchiyama Y., Nukina N., Hattori N. Propagative α-synuclein seeds as serum biomarkers for synucleinopathies. Nat Med 2023; 29(6): 1448–1455, https://doi.org/10.1038/s41591-023-02358-9.
- Manca M., Kraus A. Defining the protein seeds of neurodegeneration using real-time quaking-induced conversion assays. Biomolecules 2020; 10(9): 1233, https://doi.org/10.3390/biom10091233.
- Fairfoul G., McGuire L.I., Pal S., Ironside J.W., Neumann J., Christie S., Joachim C., Esiri M., Evetts S.G., Rolinski M., Baig F., Ruffmann C., Wade-Martins R., Hu M.T., Parkkinen L., Green A.J. Alpha-synuclein RT-QuIC in the CSF of patients with alpha-synucleinopathies. Ann Clin Transl Neurol 2016; 3(10): 812–818, https://doi.org/10.1002/acn3.338.
- Rossi M., Candelise N., Baiardi S., Capellari S., Giannini G., Orrù C.D., Antelmi E., Mammana A., Hughson A.G., Calandra-Buonaura G., Ladogana A., Plazzi G., Cortelli P., Caughey B., Parchi P. Ultrasensitive RT-QuIC assay with high sensitivity and specificity for Lewy body-associated synucleinopathies. Acta Neuropathol 2020; 140(1): 49–62, https://doi.org/10.1007/s00401-020-02160-8.
- Wang Y., Hu J., Chen X., Wang S., Zhang C., Hu J., Guo D., Liu X. Real-time quaking-induced conversion assay is accurate for Lewy body diseases: a meta-analysis. Neurol Sci 2022; 43(7): 4125–4132, https://doi.org/10.1007/s10072-022-06014-x.
- Srivastava A., Alam P., Caughey B. RT-QuIC and related assays for detecting and quantifying prion-like pathological seeds of α-synuclein. Biomolecules 2022; 12(4): 576, https://doi.org/10.3390/biom12040576.
- Grossauer A., Hemicker G., Krismer F., Peball M., Djamshidian A., Poewe W., Seppi K., Heim B. α-Synuclein seed amplification assays in the diagnosis of synucleinopathies using cerebrospinal fluid-a systematic review and meta-analysis. Mov Disord Clin Pract 2023; 10(5): 737–747, https://doi.org/10.1002/mdc3.13710.
- Brockmann K., Quadalti C., Lerche S., Rossi M., Wurster I., Baiardi S., Roeben B., Mammana A., Zimmermann M., Hauser A.K., Deuschle C., Schulte C., Waniek K., Lachmann I., Sjödin S., Brinkmalm A., Blennow K., Zetterberg H., Gasser T., Parchi P. Association between CSF alpha-synuclein seeding activity and genetic status in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol Commun 2021; 9(1): 175, https://doi.org/10.1186/s40478-021-01276-6.
- Siderowf A., Concha-Marambio L., Lafontant D.E., Farris C.M., Ma Y., Urenia P.A., Nguyen H., Alcalay R.N., Chahine L.M., Foroud T., Galasko D., Kieburtz K., Merchant K., Mollenhauer B., Poston K.L., Seibyl J., Simuni T., Tanner C.M., Weintraub D., Videnovic A., Choi S.H., Kurth R., Caspell-Garcia C., Coffey C.S., Frasier M., Oliveira L.M.A., Hutten S.J., Sherer T., Marek K., Soto C.; Parkinson’s Progression Markers Initiative. Assessment of heterogeneity among participants in the Parkinson’s progression markers initiative cohort using α-synuclein seed amplification: a cross-sectional study. Lancet Neurol 2023; 22(5): 407–417, https://doi.org/10.1016/S1474-4422(23)00109-6.
- Bongianni M., Catalan M., Perra D., Fontana E., Janes F., Bertolotti C., Sacchetto L., Capaldi S., Tagliapietra M., Polverino P., Tommasini V., Bellavita G., Kachoie E.A., Baruca R., Bernardini A., Valente M., Fiorini M., Bronzato E., Tamburin S., Bertolasi L., Brozzetti L., Cecchini M.P., Gigli G., Monaco S., Manganotti P., Zanusso G. Olfactory swab sampling optimization for α-synuclein aggregate detection in patients with Parkinson’s disease. Transl Neurodegener 2022; 11(1): 37, https://doi.org/10.1186/s40035-022-00311-3.
- Perra D., Bongianni M., Novi G., Janes F., Bessi V., Capaldi S., Sacchetto L., Tagliapietra M., Schenone G., Morbelli S., Fiorini M., Cattaruzza T., Mazzon G., Orrù C.D., Catalan M., Polverino P., Bernardini A., Pellitteri G., Valente M., Bertolotti C., Nacmias B., Maggiore G., Cavallaro T., Manganotti P., Gigli G., Monaco S., Nobili F., Zanusso G. Alpha-synuclein seeds in olfactory mucosa and cerebrospinal fluid of patients with dementia with Lewy bodies. Brain Commun 2021; 3(2): fcab045, https://doi.org/10.1093/braincomms/fcab045.
- Bargar C., Wang W., Gunzler S.A., LeFevre A., Wang Z., Lerner A.J., Singh N., Tatsuoka C., Appleby B., Zhu X., Xu R., Haroutunian V., Zou W.Q., Ma J., Chen S.G. Streamlined alpha-synuclein RT-QuIC assay for various biospecimens in Parkinson’s disease and dementia with Lewy bodies. Acta Neuropathol Commun 2021; 9(1): 62, https://doi.org/10.1186/s40478-021-01175-w.
- Stefani A., Iranzo A., Holzknecht E., Perra D., Bongianni M., Gaig C., Heim B., Serradell M., Sacchetto L., Garrido A., Capaldi S., Sánchez-Gómez A., Cecchini M.P., Mariotto S., Ferrari S., Fiorini M., Schmutzhard J., Cocchiara P., Vilaseca I., Brozzetti L., Monaco S., Jose Marti M., Seppi K., Tolosa E., Santamaria J., Högl B., Poewe W., Zanusso G; SINBAR (Sleep Innsbruck Barcelona) group. Alpha-synuclein seeds in olfactory mucosa of patients with isolated REM sleep behaviour disorder. Brain 2021; 144(4): 1118–1126, https://doi.org/10.1093/brain/awab005.
- Kuang Y., Mao H., Gan T., Guo W., Dai W., Huang W., Wu Z., Li H., Huang X., Yang X., Xu P.Y. A skin-specific α-synuclein seeding amplification assay for diagnosing Parkinson’s disease. NPJ Parkinsons Dis 2024; 10(1): 129, https://doi.org/10.1038/s41531-024-00738-7.
- Wang Z., Gilliland T., Kim H.J., Gerasimenko M., Sajewski K., Camacho M.V., Bebek G., Chen S.G., Gunzler S.A., Kong Q. A minimally invasive biomarker for sensitive and accurate diagnosis of Parkinson’s disease. Acta Neuropathol Commun 2024; 12(1): 167, https://doi.org/10.1186/s40478-024-01873-1.
- Manca M., Standke H.G., Browne D.F., Huntley M.L., Thomas O.R., Orrú C.D., Hughson A.G., Kim Y., Zhang J., Tatsuoka C., Zhu X., Hiniker A., Coughlin D.G., Galasko D., Kraus A. Tau seeds occur before earliest Alzheimer’s changes and are prevalent across neurodegenerative diseases. Acta Neuropathol 2023; 146(1): 31–50, https://doi.org/10.1007/s00401-023-02574-0.
- Saijo E., Ghetti B., Zanusso G., Oblak A., Furman J.L., Diamond M.I., Kraus A., Caughey B. Ultrasensitive and selective detection of 3-repeat tau seeding activity in Pick disease brain and cerebrospinal fluid. Acta Neuropathol 2017; 133(5): 751–765, https://doi.org/10.1007/s00401-017-1692-z.
- Saijo E., Metrick M.A. 2nd, Koga S., Parchi P., Litvan I., Spina S., Boxer A., Rojas J.C., Galasko D., Kraus A., Rossi M., Newell K., Zanusso G., Grinberg L.T., Seeley W.W., Ghetti B., Dickson D.W., Caughey B. 4-repeat tau seeds and templating subtypes as brain and CSF biomarkers of frontotemporal lobar degeneration. Acta Neuropathol 2020; 139(1): 63–77, https://doi.org/10.1007/s00401-019-02080-2.
- Kraus A., Saijo E., Metrick M.A. 2nd, Newell K., Sigurdson C.J., Zanusso G., Ghetti B., Caughey B. Seeding selectivity and ultrasensitive detection of tau aggregate conformers of Alzheimer disease. Acta Neuropathol 2019; 137(4): 585–598, https://doi.org/10.1007/s00401-018-1947-3.
- Tennant J.M., Henderson D.M., Wisniewski T.M., Hoover E.A. RT-QuIC detection of tauopathies using full-length tau substrates. Prion 2020; 14(1): 249–256, https://doi.org/10.1080/19336896.2020.1832946.
- Salvadores N., Shahnawaz M., Scarpini E., Tagliavini F., Soto C. Detection of misfolded Aβ oligomers for sensitive biochemical diagnosis of Alzheimer’s disease. Cell Rep 2014; 7(1): 261–268, https://doi.org/10.1016/j.celrep.2014.02.031.
- Estrada L.D., Chamorro D., Yañez M.J., Gonzalez M., Leal N., von Bernhardi R., Dulcey A.E., Marugan J., Ferrer M., Soto C., Zanlungo S., Inestrosa N.C., Alvarez A.R. Reduction of blood amyloid-β oligomers in Alzheimer’s disease transgenic mice by c-Abl kinase inhibition. J Alzheimers Dis 2016; 54(3): 1193–1205, https://doi.org/10.3233/JAD-151087.
- An S.S., Lim K.T., Oh H.J., Lee B.S., Zukic E., Ju Y.R., Yokoyama T., Kim S.Y., Welker E. Differentiating blood samples from scrapie infected and non-infected hamsters by detecting disease-associated prion proteins using Multimer Detection System. Biochem Biophys Res Commun 2010; 392(4): 505–509, https://doi.org/10.1016/j.bbrc.2010.01.053.
- An S.S.A., Lee B.S., Yu J.S., Lim K., Kim G.J., Lee R., Kim S., Kang S., Park Y.H., Wang M.J., Yang Y.S., Youn Y.C., Kim S. Dynamic changes of oligomeric amyloid β levels in plasma induced by spiked synthetic Aβ42. Alzheimers Res Ther 2017; 9(1): 86, https://doi.org/10.1186/s13195-017-0310-6.
- Meyer N., Janot J.M., Torrent J., Balme S. Real-time fast amyloid seeding and translocation of α-synuclein with a nanopipette. ACS Cent Sci 2022; 8(4): 441–448, https://doi.org/10.1021/acscentsci.1c01404.
- Meyer N., Bentin J., Janot J.M., Abrao-Nemeir I., Charles-Achille S., Pratlong M., Aquilina A., Trinquet E., Perrier V., Picaud F., Torrent J., Balme S. Ultrasensitive detection of Aβ42 seeds in cerebrospinal fluid with a nanopipette-based real-time fast amyloid seeding and translocation assay. Anal Chem 2023; 95(34): 12623–12630, https://doi.org/10.1021/acs.analchem.3c00017.
- Christenson P.R., Li M., Rowden G., Larsen P.A., Oh S.H. Nanoparticle-enhanced RT-QuIC (Nano-QuIC) diagnostic assay for misfolded proteins. Nano Lett 2023; 23(9): 4074–4081, https://doi.org/10.1021/acs.nanolett.3c01001.
- Kim Y., Ko S.M., Nam J.M. Protein-nanoparticle interaction-induced changes in protein structure and aggregation. Chem Asian J 2016; 11(13): 1869–1877, https://doi.org/10.1002/asia.201600236.
- Hajipour M.J., Safavi-Sohi R., Sharifi S., Mahmoud N., Ashkarran A.A., Voke E., Serpooshan V., Ramezankhani M., Milani A.S., Landry M.P., Mahmoudi M. An overview of nanoparticle protein corona literature. Small 2023; 19(36): e2301838, https://doi.org/10.1002/smll.202301838.
- Chatzikonstantinou S., Kazis D., Karantali E., Knights M., McKenna J., Petridis F., Mavroudis I. A meta-analysis on RT-QuIC for the diagnosis of sporadic CJD. Acta Neurol Belg 2021; 121(2): 341–349, https://doi.org/10.1007/s13760-021-01596-3.
- Krawczuk D., Groblewska M., Mroczko J., Winkel I., Mroczko B. The role of α-synuclein in etiology of neurodegenerative diseases. Int J Mol Sci 2024; 25(17): 9197, https://doi.org/10.3390/ijms25179197.
- Zhang K., Mizuma H., Zhang X., Takahashi K., Jin C., Song F., Gao Y., Kanayama Y., Wu Y., Li Y., Ma L., Tian M., Zhang H., Watanabe Y. PET imaging of neural activity, β-amyloid, and tau in normal brain aging. Eur J Nucl Med Mol Imaging 2021; 48(12): 3859–3871, https://doi.org/10.1007/s00259-021-05230-5.
- Park M.C., Kim M., Lim G.T., Kang S.M., An S.S., Kim T.S., Kang J.Y. Droplet-based magnetic bead immunoassay using microchannel-connected multiwell plates (μCHAMPs) for the detection of amyloid beta oligomers. Lab Chip 2016; 16(12): 2245–2253, https://doi.org/10.1039/c6lc00013d.
- Cheong D.Y., Lee W., Park I., Park J., Lee G. Amyloid formation in nanoliter droplets. Int J Mol Sci 2022; 23(10): 5480, https://doi.org/10.3390/ijms23105480.
- Pfammatter M., Andreasen M., Meisl G., Taylor C.G., Adamcik J., Bolisetty S., Sánchez-Ferrer A., Klenerman D., Dobson C.M., Mezzenga R., Knowles T.P.J., Aguzzi A., Hornemann S. Absolute quantification of amyloid propagons by digital microfluidics. Anal Chem 2017; 89(22): 12306–12313, https://doi.org/10.1021/acs.analchem.7b03279.








